 |
Composición:
Cada jeringa prellenada contiene: Denosumab 60 mg, en una solución de 1.0 ml (60 mg/ml). Lista de Excipientes: Acetato; Hidróxido de Sodio; Sorbitol; Polisorbato 20; Agua para Inyecciones. Presentación farmacéutica: Solución para inyección S.C. Solución transparente, de incolora a ligeramente amarilla, con un pH de 5.2 y que puede contener oligoelementos proteicos translúcidos o de color blanco.
|
|
Indicaciones:
Osteoporosis postmenopáusica: Prolia está indicado en el tratamiento de la osteoporosis en mujeres postmenopáusicas. En mujeres postmenopáusicas que padecen osteoporosis, Prolia aumenta la densidad mineral ósea (BMD) y disminuye la incidencia de fracturas de cadera, vertebrales y no vertebrales. Pérdida ósea en pacientes sometidos a terapia antineoplásica con ablación hormonal: Prolia está indicado en el tratamiento de la pérdida ósea en pacientes sometidos a terapia con ablación hormonal para cáncer prostático o mamario. En pacientes que padecen cáncer prostático, Prolia disminuye la incidencia de fracturas vertebrales.
|
|
Propiedades:
Farmacodinamia: Mecanismo de acción: El Denosumab es un anticuerpo monoclonal humano (IgG2) que se dirige y fija con gran afinidad y especificidad al ligando RANKL, evitando que éste active su único receptor, el RANK, en la superficie de los osteoclastos y sus precursores, que es independiente de la superficie ósea. La prevención de la interacción RANKL /RANK inhibe la formación, función y supervivencia de los osteoclastos. Por lo tanto, el Denosumab reduce la reabsorción ósea e incrementa la masa y la fuerza óseas, tanto en los huesos corticales como en los trabeculares. Efectos farmacodinámicos: En estudios clínicos, el tratamiento con 60 mg de Denosumab produjo una rápida reducción en el nivel de marcadores séricos de reabsorción ósea, C-telopéptidos (CTX) tipo 1, dentro de las 6 horas posteriores a la administración subcutánea (en aproximadamente 70%), exhibiendo reducciones de aproximadamente 85% a los 3 días. Las reducciones en los niveles de CTX fueron constantes durante el intervalo posológico de 6 meses. Al final de cada intervalo posológico, las reducciones en los niveles de CTX se vieron atenuadas parcialmente desde la reducción máxima de ł 87% hasta aproximadamente ł 45% (intervalo de 45-80%), lo cual refleja la reversibilidad de los efectos del Denosumab en el remodelado óseo una vez que disminuyen sus niveles séricos. Estos efectos pudieron mantenerse con la continuación del tratamiento. De manera consistente con el acoplamiento fisiológico de la formación y la reabsorción de los huesos en el remodelado esquelético, se observaron reducciones en los niveles de marcadores de formación ósea (p.ej., fosfatasa alcalina específica a huesos [BSAP, por sus siglas en inglés] y fragmento N-terminal del propéptido sérico de colágeno tipo 1 [P1NP, por sus siglas en inglés]) al inicio del primer mes posterior a la administración de la primera dosis de Denosumab. Los marcadores de recambio óseo (marcadores de reabsorción y formación óseas) generalmente alcanzaron los niveles pretratamiento dentro de los 9 meses posteriores a la administración de la última dosis subcutánea de 60 mg. Al volver a iniciar la administración, el grado de inhibición del CTX por parte del Denosumab fue similar al observado en los pacientes que iniciaron un tratamiento con Denosumab. Inmunogenicidad: El Denosumab es un anticuerpo monoclonal humano; al igual que con todas las proteínas terapéuticas, existe un riesgo teórico de inmunogenicidad. Más de 13.000 pacientes fueron sometidos a un escrutinio sobre anticuerpos de fijación, utilizando un inmunoensayo de electroquimioluminiscencia sensible por puenteo. Menos de 1% de los pacientes tratados con Denosumab hasta por 5 ańos mostraron resultados positivos (incluyendo anticuerpos preexistentes, transitorios y en desarrollo). Los pacientes que mostraron resultados positivos en cuanto a anticuerpos de fijación fueron evaluados posteriormente en cuanto a anticuerpos neutralizantes, utilizando un ensayo biológico in vitro de quimioluminiscencia basado en células, y ninguno de ellos mostró resultados positivos. No hubo indicios de alteración en el perfil farmacocinético, el perfil de toxicidad o la respuesta clínica que estuviera asociada con el desarrollo de anticuerpos de fijación. Farmacocinética: Después de su administración subcutánea, el Denosumab mostró una farmacocinética no lineal con la dosis a lo largo de un amplio intervalo posológico, así como incrementos proporcionales a la dosis en el grado de exposición a dosis de 60 mg (ó 1 mg/kg) y mayores. Absorción: Después de administrar una dosis subcutánea de 60 mg de Denosumab, su biodisponibilidad fue de 61% y se alcanzaron concentraciones séricas máximas de Denosumab (Cmax) de 6 µg/ml (intervalo de 1-17 µg /mL) en 10 días (intervalo de 2-28 días). Después de lograr la Cmax, los niveles séricos declinaron con una vida media de 26 días (intervalo de 6-52 días) a lo largo de un periodo de 3 meses (intervalo de 1.5-4.5 meses). El 53% de los pacientes no mostró cantidades cuantificables de Denosumab a los 6 meses posteriores a la dosis. Distribución: Al administrar de manera subcutánea dosis múltiples de 60 mg una vez cada 6 meses, no se observó acumulación o cambio alguno en la farmacocinética del Denosumab a través del tiempo. Metabolismo: El Denosumab está compuesto únicamente de aminoácidos y carbohidratos como la inmunoglobulina nativa. Con base en datos no clínicos, se espera que el metabolismo del Denosumab siga las vías de depuración de la inmunoglobulina, dando como resultado su degradación en pequeńos péptidos y aminoácidos individuales. Eliminación: El Denosumab está compuesto únicamente de aminoácidos y carbohidratos como la inmunoglobulina nativa, por lo cual no se espera que sea eliminado a través de mecanismos metabólicos hepáticos (p.ej., las enzimas del citocromo p450 (CYP)). Con base en datos no clínicos, se espera que su eliminación siga las vías de depuración de la inmunoglobulina, dando como resultado su degradación en pequeńos péptidos y aminoácidos individuales. Poblaciones de pacientes especiales: Pacientes de edad avanzada (de 65 ańos de edad o mayores): En un análisis farmacocinético poblacional de pacientes cuyas edades variaron de los 28 a los 87 ańos de edad, no se encontró que la edad fuera un factor significativo en la farmacocinética del Denosumab. Nińos y adolescentes (de hasta 18 ańos de edad): No se dispone de datos farmacocinéticos en pacientes pediátricos. Raza: La farmacocinética del Denosumab no se vio afectada por la raza en mujeres postmenopáusicas o en pacientes de cáncer de mama sometidas a terapia de ablación hormonal. Insuficiencia renal: En un estudio realizado en 55 pacientes con grados variables de función renal, incluyendo pacientes sometidos a diálisis, el grado de insuficiencia renal no produjo efecto alguno en la farmacocinética y la farmacodinamia del Denosumab; por lo tanto, no es necesario ajustar la dosis en presencia de insuficiencia renal. Insuficiencia hepática: No se han realizado estudios clínicos para evaluar el efecto de la insuficiencia hepática en la farmacocinética del Denosumab. Estudio clínico: Tratamiento de osteoporosis postmenopáusica: Se demostraron los perfiles de seguridad y eficacia del Denosumab en el tratamiento de la osteoporosis postmenopáusica en el estudio FREEDOM, un estudio multinacional, doble ciego, aleatorizado, controlado con placebo y de 3 ańos de duración, que demostró que el Denosumab era eficaz al compararse con placebo en cuanto a la reducción de la incidencia de nuevas fracturas de cadera, vertebrales y no vertebrales en mujeres postmenopáusicas con osteoporosis. Se reclutó a 7,808 mujeres de 60-91 ańos de edad, de las cuales el 23.6% tenía fracturas vertebrales prevalentes. Las mujeres fueron distribuidas aleatoriamente para recibir inyecciones subcutáneas de placebo (n=3.906), o bien, 60 mg de Denosumab (n=3.902), una vez cada 6 meses. Las mujeres recibieron complementos diarios de calcio (cuando menos 1.000 mg) y vitamina D (cuando menos 400 IU). La variable primaria de eficacia fue la incidencia de nuevas fracturas vertebrales. Las variables secundarias de eficacia incluyeron la incidencia de fracturas no vertebrales y fracturas de cadera, evaluadas a los 3 ańos. En este estudio, la terapia con Denosumab redujo significativamente el riesgo de desarrollar nuevas fracturas vertebrales, no vertebrales y de cadera, en comparación con el placebo. Los 3 criterios de valoración de eficacia por fracturas lograron el nivel de significancia estadística basado en el esquema preespecificado de pruebas secuenciales. Efecto en las fracturas vertebrales: El Denosumab redujo significativamente el riesgo de desarrollar nuevas fracturas vertebrales (criterio primario de valoración) en 68% (cociente de riesgo: 0.32; p<0.0001) a lo largo de 3 ańos. Las tasas de fracturas en 3 ańos correspondientes a nuevas fracturas vertebrales fueron de 7.2% en el grupo placebo y de 2.3% en el grupo tratado con Prolia (reducción en el riesgo absoluto no ajustado de 4,8%). También se observaron reducciones a lo largo de 1 ańo (reducción en el riesgo relativo de 61%; reducción en el riesgo absoluto no ajustado de 1.4%) y a lo largo de 2 ańos (reducción en el riesgo relativo de 71%; reducción en el riesgo absoluto no ajustado de 3.5) (todos los valores p < 0.0001). El Denosumab también redujo el riesgo de desarrollar fracturas pertenecientes a otras categorías preespecificadas, incluyendo nuevas fracturas vertebrales y el agravamiento de las ya existentes (reducción en el riesgo relativo de 67%, reducción en el riesgo absoluto no ajustado de 4.8%), nuevas fracturas vertebrales múltiples (reducción en el riesgo relativo de 61%, reducción en el riesgo absoluto no ajustado de 1.0%), fracturas vertebrales clínicas (reducción en el riesgo relativo de 69%, reducción en el riesgo absoluto no ajustado de 1.8%) a lo largo de 3 ańos. Las reducciones en el riesgo de desarrollar nuevas fracturas vertebrales, como consecuencia de la administración de Denosumab a lo largo de 3 ańos, fueron consistentes y significativas, independientemente del riesgo basal de desarrollar fracturas osteoporóticas mayores en 10 ańos, de acuerdo a la evaluación de FRAX® (algoritmo de la WHO para la Evaluación del Riesgo de desarrollar Fracturas), y de si las mujeres tenían alguna fractura vertebral prevalente o antecedentes de fracturas no vertebrales, e independientemente de la edad basal, BMD, nivel de recambio óseo y uso previo de algún medicamento para la osteoporosis. En mujeres postmenopáusicas con osteoporosis mayores de 75 ańos de edad, el Denosumab redujo la incidencia de desarrollar nuevas fracturas vertebrales (64%) y no vertebrales (16%). Efecto en las fracturas clínicas: El Denosumab redujo significativamente el riesgo de desarrollar fracturas no vertebrales (criterio de valoración secundario) en 20% (cociente de riesgo: 0.80; p = 0.0106) a lo largo de 3 ańos. Las tasas de fracturas en 3 ańos correspondientes a fracturas no vertebrales fueron de 8.0% en el grupo placebo y de 6.5% en el grupo tratado con Denosumab (reducción en el riesgo absoluto no ajustado de 1.5%). El Denosumab también redujo el riesgo de desarrollar fracturas clínicas (reducción en el riesgo relativo de 30%, reducción en el riesgo absoluto no ajustado de 2.9%), fracturas no vertebrales mayores (reducción en el riesgo relativo de 20%, reducción en el riesgo absoluto no ajustado de 1.2%) y fracturas osteoporóticas mayores (reducción en el riesgo relativo de 35%, reducción en el riesgo absoluto no ajustado de 2.7%) a lo largo de 3 ańos. En mujeres con una puntuación-T basal de BMD en el cuello femoral 2.5, el Denosumab redujo la incidencia de fracturas no vertebrales (reducción en el riesgo relativo de 35%, reducción en el riesgo absoluto no ajustado de 4.1%, p < 0.001) a lo largo de 3 ańos. Se observaron reducciones en la incidencia de fracturas no vertebrales independientemente de la probabilidad basal de desarrollar alguna fractura osteoporótica mayor en 10 ańos, de acuerdo a la evaluación de FRAX®. Efecto en las fracturas de cadera: El Denosumab redujo significativamente el riesgo de desarrollar fracturas de cadera (criterio secundario de valoración) en 40% (cociente de riesgo: 0.60; p = 0.0362) a lo largo de 3 ańos. Las tasas de fracturas en 3 ańos correspondientes a fracturas de cadera fueron de 1.2% en el grupo placebo y de 0.7% en el grupo tratado con Denosumab (reducción en el riesgo absoluto no ajustado de 0.5%). Las reducciones en el riesgo de desarrollar fracturas de cadera a lo largo de 3 ańos fueron consistentes y significativas, independientemente de la probabilidad basal de desarrollar alguna fractura de cadera en 10 ańos, de acuerdo a la evaluación de FRAX®. En mujeres que se encuentran en mayor riesgo de desarrollar fracturas, de acuerdo a lo definido anteriormente por la edad basal, BMD y fractura vertebral prevalente, se observó una reducción en el riesgo relativo de 48% con Denosumab (reducción en el riesgo absoluto no ajustado 1.1%). En un análisis post-hoc realizado en mujeres postmenopáusicas con osteoporosis que se encontraban alrededor de los 75 ańos de edad, el Denosumab redujo la incidencia de fracturas de cadera (62%). Efecto en la densidad mineral ósea (BMD, por sus siglas en inglés): El Denosumab incrementó la BMD en todos los sitios clínicos cuantificados, en relación con el tratamiento con placebo a 1, 2 y 3 ańos. El Denosumab incrementó la BMD en 9.2% en la espina lumbar, 6.0% en la cadera total, 4.8% en el cuello femoral, 7.9% en el trocánter de la cadera, 3.5% en el radio distal 1/3 y 4.1% en el cuerpo en general, a lo largo de 3 ańos. Los incrementos en la BMD en la espina lumbar, la cadera total y el trocánter de la cadera se observaron en el primer mes posterior a la administración de la dosis inicial. El Denosumab incrementó la BMD de la espina lumbar en 96% con respecto a la línea basal en mujeres postmenopáusicas a los 3 ańos. Se observaron efectos consistentes en la BMD de la espina lumbar, independientemente de la edad basal, la raza, el peso/BMI, la BMD y el nivel de recambio óseo. Histología de los Huesos: Las evaluaciones histológicas mostraron que los huesos eran de arquitectura y calidad normales, y demostraron la disminución esperada en el nivel de recambio óseo con respecto al tratamiento con placebo. No hubo indicios de defectos de mineralización, hueso no laminar o fibrosis medular. Eficacia clínica en el tratamiento de la pérdida ósea asociada con terapia de ablación hormonal: Tratamiento de la pérdida ósea asociada con la privación de andrógenos: Se evaluaron los perfiles de eficacia y seguridad del Denosumab en el tratamiento de la pérdida ósea asociada con la privación de andrógenos, en un estudio multinacional, aleatorizado, doble ciego, controlado con placebo y de 3 ańos de duración, realizado en 1.468 con cáncer de próstata no metastásico de 48-97 ańos de edad. Los hombres menores de 70 ańos de edad también tenían una puntuación-T de BMD en la espina lumbar, la cadera total o el cuello femoral <- 1.0, o bien, antecedentes de alguna fractura osteoporótica. Los sujetos recibieron inyecciones subcutáneas de 60 mg de Denosumab (n=734) o placebo (n=734), una vez cada 6 meses. Los hombres recibieron complementación diaria de calcio (cuando menos 1.000 mg) y vitamina D (cuando menos 400 IU). Se observaron incrementos significativos en la BMD de la espina lumbar, la cadera total, el cuello femoral y el trocánter de la cadera en el primer mes posterior a la administración de la dosis inicial. El Denosumab incrementó BMD de espina lumbar en 7.9%, la BMD de la cadera total en 5.7%, la BMD del cuello femoral en 4.9%, la BMD del trocánter de la cadera en 6.9%, la BMD del radio distal 1/3 en 6.9%, y la BMD del cuerpo en general en 4.7% a lo largo de 3 ańos, en comparación con el placebo (P < 0,0001). Se observaron efectos consistentes en la BMD de la espina lumbar, independientemente de la edad, la raza, la región geográfica, el peso /BMI, la BMD y el nivel de recambio óseo; la duración de la privación de andrógenos y la presencia de fracturas vertebrales en la línea basal. El Denosumab redujo significativamente el riesgo de desarrollar nuevas fracturas vertebrales en 62% (cociente de riesgo: 0,38; p<0.0063) a lo largo de 3 ańos. También se observaron reducciones a lo largo de 1 ańo (reducción en el riesgo relativo de 85%; reducción en el riesgo absoluto de 1,6%), y a los 2 ańos (reducción en el riesgo relativo de 69%; reducción en el riesgo absoluto de 2.2%) (todos los valores p < 0.01). El Denosumab también redujo la incidencia por sujeto de más de una fractura osteoporótica en cualquier sitio en 72%, en relación con el placebo, a lo largo de 3 ańos (placebo 2.5% vs. Denosumab 0.7%, p = 0.0063). Tratamiento de la pérdida ósea en mujeres sometidas a terapia con inhibidores de la aromatasa para el cáncer de mama: Se evaluaron los perfiles de eficacia y seguridad del Denosumab en el tratamiento de la pérdida ósea asociada con la terapia ayudante con inhibidores de la aromatasa, en un estudio multinacional, aleatorizado, doble ciego, controlado con placebo y de 2 ańos de duración, realizado en 252 mujeres con cáncer de mama no metastásico de 35-84 ańos de edad. Las mujeres tienen puntuaciones-T basales de BMD entre -1.0 a -2.5 en la espina lumbar, la cadera total o el cuello femoral. Las mujeres fueron distribuidas aleatoriamente para recibir inyecciones subcutáneas de 60 mg de Denosumab (n=127), o bien, de placebo (n=125), una vez cada 6 meses. Las mujeres recibieron complementación diaria de calcio (cuando menos 1.000 mg) y vitamina D (cuando menos 400 IU). La variable primaria de eficacia fue el cambio porcentual en la BMD de la espina lumbar. El Denosumab incrementó la BMD en todos los sitios clínicos cuantificados, en relación con el tratamiento con placebo a 2 ańos: 7.6% en la espina lumbar, 4.7% en la cadera total, 3.6% en el cuello femoral, 5.9% en el trocánter de la cadera, 6.1% en el radio distal 1/3 y 4.2% en el cuerpo en general. Se observaron incrementos significativos en la BMD de la espina lumbar en el primer mes posterior a la administración de la dosis inicial. Se observaron efectos consistentes en la BMD de la espina lumbar, independientemente de la edad basal, la duración de la terapia con inhibidores de la aromatasa, el peso/BMI, el uso previo de quimioterapia, el uso previo de moduladores selectivos de receptores de estrógenos (SERM, por sus siglas en inglés), y el tiempo transcurrido desde la menopausia. Datos preclínicos de seguridad: Carcinogenicidad: Aún no se evalúa el potencial carcinogénico del Denosumab en estudios a largo plazo realizados en animales. Mutagenicidad: Aún no se evalúa el potencial genotóxico del Denosumab. Toxicología en la reproducción: Fertilidad: El Denosumab no produjo efecto alguno en la fertilidad femenina ni en los órganos reproductivos de monos machos, a niveles de exposición AUC que fueron de 100 a 150 veces superiores al nivel de exposición en humanos que se logra al administrar subcutáneamente una dosis de 60 mg una vez cada 6 meses. Embarazo: A niveles de exposición hasta 100 veces superiores al nivel de exposición en humanos, el Denosumab no mostró indicios de afectar al feto en estudios de toxicidad en el desarrollo. Farmacología en animales: El tratamiento a largo plazo (16 meses) de monos hembras ovariectomizadas, con dosis SC de Denosumab de 25 ó 50 mg/kg administradas una vez al mes, se asoció con incrementos significativos en la masa, la densidad (BMD) y la fuerza de los huesos esponjosos y corticales. El tejido fue normal, sin indicios de defectos de mineralización, acumulación de osteoides o hueso no laminar. La transición del tratamiento durante 6 meses con Alendronato a 25 mg/kg de Denosumab en monos hembras ovariectomizadas no ocasionó ninguna disminución significativa en los niveles séricos de calcio. Se mantuvieron o se mejoraron la fuerza y la reducción óseas en la reabsorción ósea de todos los sitios esqueléticos. Se observaron placas con crecimiento anormal en monos adolescentes que recibieron dosis SC de Denosumab de 10 y 50 mg/kg (27 y 150 veces el nivel de exposición AUC en adultos humanos que reciben una dosis SC de Denosumab de 60 mg cada 6 meses), lo cual es consistente con la actividad farmacológica del Denosumab. A partir de ratones alterados genéticamente (noqueados) que carecen del ligando RANK o RANKL, y mediante el uso de inhibidores de la vía RANKL en roedores, como la OPG-Fc, se ha obtenido información adicional sobre las propiedades farmacodinámicas del Denosumab. Ratones noqueados: (1) tienen ausencia de lactancia debido a la inhibición de la maduración de las glándulas mamarias (desarrollo de la glándula lobuloalveolar durante el embarazo); (2) exhibían un deterioro en la formación de los nódulos linfáticos; y (3) exhibían una reducción en el crecimiento óseo y una falta de erupción de dientes. Se observaron cambios fenotípicos similares en un estudio corroborativo que se realizó en ratas de 2 semanas de edad que recibieron OPG-Fc. Los estudios de distribución histológica indicaron que el Denosumab no se fija a tejidos conocidos por expresar a otros miembros de la superfamilia de TNF, incluyendo el ligando inductor de apoptosis que se relaciona con TNF (TRAIL, por sus siglas en inglés).
|
|
Posología:
Administración: La administración debe efectuarla un individuo que haya sido capacitado adecuadamente en cuanto a técnicas de inyectar. Dosis: La dosis recomendada de Prolia consiste en 1 sola inyección subcutánea de 60 mg, administrada 1 vez cada 6 meses. Los pacientes deben recibir complementos de calcio y vitamina D mientras se encuentren bajo tratamiento. Poblaciones: Nińos: Prolia no se recomienda pacientes pediátricos, ya que aún no se establecen los perfiles de seguridad y eficacia de Prolia en el grupo de edad pediátrica. En estudios realizados en animales, se ha acoplado la inhibición del ligando RANK/RANK (RANKL), con una construcción de osteoprotegerina fijada a Fc (OPG-Fc), a la inhibición del crecimiento óseo y la falta de erupción de dientes (véase Datos Preclínicos de Seguridad). Por lo tanto, el tratamiento con Denosumab es capaz de afectar el crecimiento de los huesos en nińos con placas epifisiarias abiertas, así como de inhibir la erupción de los dientes. Pacientes de edad avanzada: De acuerdo a los datos disponibles sobre seguridad y eficacia en pacientes de edad avanzada, no se requiere ajustar la dosificación (véase Farmacocinética: Poblaciones de pacientes especiales). Insuficiencia renal: De acuerdo a los datos disponibles sobre seguridad y eficacia en pacientes de edad avanzada, no se requiere ajustar la dosificación en pacientes con insuficiencia renal (véase Farmacocinética: Poblaciones de pacientes especiales). Los pacientes con insuficiencia renal grave (depuración de creatinina < 30 ml/min) o sometidos a diálisis se encuentran en mayor riesgo de desarrollar hipocalcemia. Es importante que se instituya una ingesta adecuada de calcio y vitamina D en pacientes con insuficiencia renal grave o que reciban diálisis. Insuficiencia hepática: Aún no se han estudiado los perfiles de seguridad y eficacia de Prolia en pacientes con insuficiencia hepática.
|
|
Modo de Empleo:
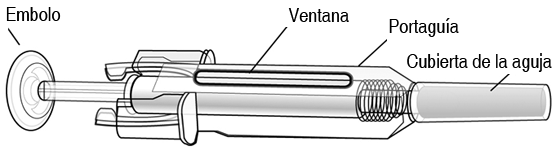
Las personas sensibles al látex no deben manipular el tapón de la aguja que se encuentra en la jeringas prellenadas para uso único, el cual contiene caucho natural seco (un derivado del látex). Antes de su administración, se debe inspeccionar la solución de Prolia en cuanto a presencia de partículas y decoloración. La solución no debe utilizarse si se encuentra turbia o decolorada. No agitar. Para evitar malestares en el sitio de la inyección, permita que la jeringa prellenada alcance la temperatura ambiente (hasta 25°C) antes de la inyección, e inyecte lentamente. Inyecte todo el contenido de la jeringa prellenada. Deseche cualquier medicamento remanente en la jeringa prellenada. En el folleto de información al paciente se incluyen las instrucciones para la autoadministración a través de una inyección subcutánea. Se deberá desechar cualquier producto no utilizado o material de desecho, de conformidad con los requisitos locales. No todas las presentaciones se encuentran disponibles en todos los países. Instrucciones para aplicar una inyección con la jeringa prellenada de prolia con un portaguja automático: Esta sección contiene información sobre el uso de la jeringa prellenada de Prolia. Es importante que usted o su cuidador no apliquen las inyecciones, a menos que hayan recibido capacitación por parte de su médico o proveedor de servicios de salud. Siempre lave sus manos antes de aplicar cualquier inyección. Si tiene alguna pregunta sobre cómo aplicar la inyección, favor de solicitar la asistencia de su médico o proveedor de servicios de salud.
Ver Tabla
Antes de comenzar: Lea cuidadosamente todas las instrucciones antes de utilizar la jeringa prellenada. Para reducir el riesgo de punciones accidentales con la aguja en los usuarios, cada jeringa prellenada viene con un portaguja que se activa automáticamente para cubrir la aguja después de haber completado la entrega del contenido de la jeringa prellenada. No intenté activar el portaguja antes de la inyección. No utilice la jeringa prellenada si la cubierta de la aguja ha sido retirada, o si el portaguja ha sido activado (cubriendo la aguja). żCómo utilizar la jeringa prellenada de Prolia?: Su médico le ha prescrito una jeringa prellenada de Prolia para ser inyectada en el tejido que se encuentra justo debajo de la piel (subcutáneo). Usted debe inyectar todo el contenido (1 ml) de la jeringa prellenada de Prolia, y deberá hacerlo una vez cada 6 meses, de acuerdo a las indicaciones de su médico o proveedor de servicios de salud. Equipo: Para aplicar una inyección, usted necesitará: 1. Una nueva jeringa prellenada de Prolia; y 2. Toallitas o algún producto similar con alcohol. żQué hacer antes de administrar una inyección subcutánea de Prolia?: 1. Retire la jeringa prellenada del refrigerador. No tome la jeringa prellenada por el émbolo o la cubierta de la aguja. Esto podría dańar el dispositivo. 2. La jeringa prellenada puede dejarse fuera del refrigerador para que alcance la temperatura ambiente. Esto hará que la inyección sea más cómoda. No la caliente de alguna otra forma; por ejemplo, en un horno de microondas o en agua caliente. No deje la jeringa expuesta a la luz directa. 3. No agite la jeringa prellenada excesivamente. 4. No retire la cubierta de la aguja de la jeringa prellenada hasta que esté listo para aplicar la inyección. 5. Verifique la fecha de caducidad en la etiqueta de la jeringa prellenada (EXP:). No la utilice si la fecha ha excedido el último día del mes mostrado. 6. Verifique la apariencia de Prolia. Debe ser una solución transparente, de incolora a ligeramente amarilla. La solución no debe ser inyectada si está turbia o decolorada. 7. Busque una superficie limpia, cómoda y libre para colocar todo el equipo a su alcance. 8. Lave bien sus manos. żDónde se debe aplicar la inyección?: A. Los mejores lugares para aplicar la inyección son la parte superior de sus muslos y el abdomen. Su cuidador también puede utilizar el área externa de sus antebrazos.
Ver Tabla
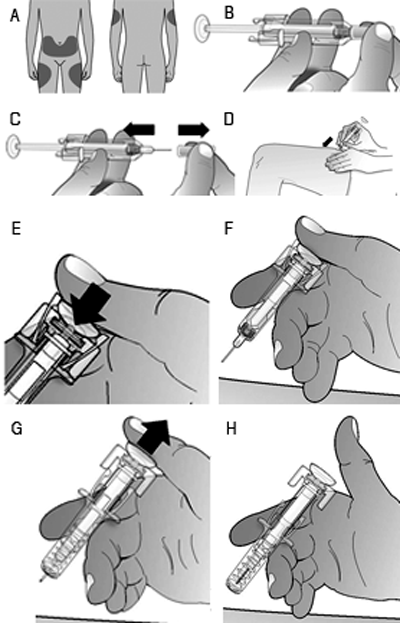
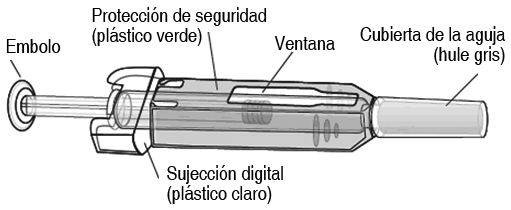
żCómo aplicar la inyección?: 1. Desinfecte la piel utilizando una toallita con alcohol. 2. B. Para evitar que se doble la aguja, jale suavemente la cubierta de la misma hacia afuera sin torcerla, tal como se muestra la imagen. No toque la aguja ni presione el émbolo.1. Es posible que se observe una pequeńa burbuja en la jeringa prellenada. No tiene que retirar la burbuja de aire antes de aplicar la inyección. La inyección de la solución con la burbuja de aire no produce dańo alguno. 2. C. Haga un pliegue (sin exprimir) en la piel entre sus dedos pulgar e índice. Introduzca completamente la aguja la piel, siguiendo las indicaciones de su médico o proveedor de servicios de salud. 5. D. Presione el émbolo con una presión constante y lenta, sin dejar de hacer el pliegue en la piel. Presione el émbolo hasta el fondo para inyectar toda la solución. El portaguja no se activará a menos que usted vacíe la jeringa prellenada. 6. E. F. G. H. Mientras mantiene presionado el émbolo, retire la aguja y suelte la piel. Retire el émbolo y permita que la jeringa se mueva hasta que toda la aguja está cubierta por el portaguja. 7. Si el portaguja no se activa, es posible que se haya aplicado una inyección incompleta. Llame a su médico o proveedor de servicios de salud si usted cree que no ha recibido toda la dosis. No vuelva a colocar la cubierta de la aguja en jeringas usadas. 8. Si observa una mancha de sangre, puede limpiarla suavemente con una mota o tejido de algodón. No frote el sitio de inyección. Si fuera necesario, usted puede cubrir el sitio de la inyección con un parche. 9. Utilice cada jeringa prellenada solamente para una inyección. No utilice cualquier parte remanente de Prolia en la jeringa. Recuerde: si tiene algún problema, solicite la ayuda y la asesoría de su médico. Desecho de jeringas utilizadas: NO vuelva colocar la cubierta de la aguja en jeringas usadas. Conserve las jeringas usadas fuera del alcance y la vista de los nińos. La jeringa usada debe ser desechada de conformidad con los requisitos locales. Pregúntele a su farmacéutico cómo deshacerse de los medicamentos que ya no requiera. Estas medidas ayudan a proteger el ambiente. Instrucciones para aplicar un inyección con la jeringa prellenada de prolia con un portaguja manual: Importante: a fin de minimizar las punciones accidentales con la aguja, la jeringa prellenada para uso único de Prolia tendrá una protección de seguridad de color verde; active manualmente la protección de seguridad después de aplicar la inyección. No deslice la protección de seguridad de color verde sobre la aguja antes administrar la inyección; se bloqueará el dispositivo y evitará la inyección.
Ver Tabla
Active la protección de seguridad de color verde (deslícela sobre la aguja) después de aplicar la inyección. La tapa de la aguja de color gris en la jeringa prellenada para uso único contiene caucho natural seco (un derivado del látex); las personas sensibles al látex no deben manipular la tapa.
Ver Tabla
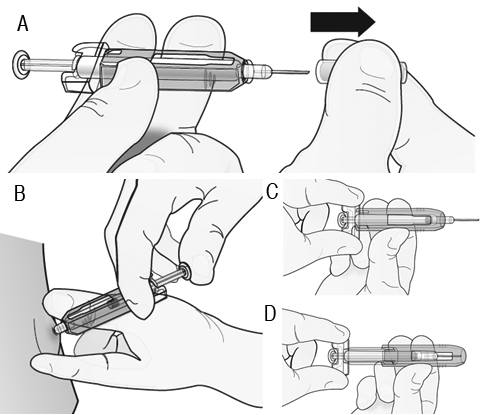
Paso 1: retire la tapa de la aguja de color gris. Paso 2: administre la inyección. NO vuelva a colocar la tapa de la aguja de color gris en la aguja. Paso 3: deslice inmediatamente la protección de seguridad de color verde sobre la aguja. Con la aguja apuntando lejos de usted: Sostenga la jeringa prellenada por el agarre de plástico transparente para los dedos con una mano. Después, con la otra mano, tome la protección de seguridad de color verde por su base y deslícela suavemente hacia la aguja hasta que quede asegurada en su lugar y/o usted escuche un "clic." NO tome la protección de seguridad de color verde con mucha firmeza - se moverá fácilmente si usted la sostiene y la desliza suavemente. Deseche inmediatamente la jeringa y la tapa de la aguja en el contenedor de agujas más cercano. No vuelva a colocar la tapa de la aguja en la jeringa usada.
|
|
Efectos Colaterales:
Datos de estudios clínicos: A continuación se enumeran las reacciones adversas por clase de sistema de órganos corporales según la MedDRA, así como por frecuencia. A continuación se presentan las categorías de frecuencia basadas en tasas de eventos ocurridos durante 1 ańo: Muy común ł 1 en 10; ł Común 1 en 100 y < 1 en 10; No común ł 1 en 1.000 y < 1 en 100; Rara ł 1 en 10.000 y < 1 en 1.000; Muy rara < 1/10.000. Dentro de cada agrupación de frecuencia y clase de sistema de órganos, se presentan los efectos adversos en orden de gravedad decreciente. Infecciones e infestaciones: Común: Infección del tracto urinario. Infección de tracto respiratorio superior. No común: Diverticulitis1. Infección del oído. Celulitis. Trastornos metabólicos y nutricionales: Muy raras: Hipocalcemia1. Trastornos del sistema nervioso: Común: Ciática. Trastornos oculares: Cataratas2. Trastornos gastrointestinales: Común: Estreńimiento. Trastornos de la piel y del tejido subcutáneo: No común: Eccema3. Común: Erupción cutánea. Trastornos musculoesqueléticos y del tejido conjuntivo: Común: Dolor en las extremidades. Rara: Osteonecrosis mandibular. 1Véase Advertencias y Precauciones. 2 En hombres con cáncer de próstata que reciben terapia con privación de andrógenos. 3 Incluye dermatitis, dermatitis alérgica, dermatitis atópica y dermatitis por contacto.
|
|
Contraindicaciones:
Hipocalcemia.
|
|
Advertencias:
Es importante que se instituya una ingesta adecuada de calcio y vitamina D en todos los pacientes que reciban Prolia. Se debe corregir la hipocalcemia a través de una ingesta adecuada de calcio y vitamina D antes de iniciar la terapia. Se recomienda instituir una vigilancia clínica de los niveles de calcio en aquellos patentes predispuestos a padecer hipocalcemia (véase Efectos Colaterales). Los pacientes bajo tratamiento con Prolia podrían desarrollar infecciones cutáneas (predominantemente celulitis) que conduzcan a su hospitalización. Estos eventos fueron comunicados con más frecuencia en el grupo tratado con Denosumab (0.4%) que en el grupo tratado con placebo (0.1%). (Véase Efectos colaterales). La incidencia general de las infecciones cutáneas fue similar entre los grupos tratados con placebo y Denosumab. Se debe aconsejar a los pacientes que busquen atención médica oportuna si desarrollan signos o síntomas de celulitis. Se han comunicado casos de osteonecrosis mandibular (ONM) de manera predominante en pacientes con cáncer avanzado bajo tratamiento con 120 mg cada 4 semanas. Se ha comunicado rara vez la ONM en pacientes con osteoporosis bajo tratamiento con 60 mg cada 6 meses (véase Efectos colaterales). Los factores de riesgo conocidos de la ONM incluyen el diagnóstico de cáncer con lesiones óseas, los tratamientos concomitantes (p. ej., Quimioterapia, medicamentos biológicos antiangiogénicos, corticosteroides, radioterapia de cabeza y cuello), una higiene bucal deficiente, extracciones dentales y comorbilidades (p. ej., enfermedad dental preexistente, amemia, coagulopatía, infección) y tratamiento previo con biofosfonatos. En pacientes con factores de riesgo concomitantes se debe considerar la realización de una revisión dental con un tratamiento odontológico preventivo apropiado antes de iniciar el tratamiento con Prolia. Durante el tratamiento, si es posible, estos pacientes deben evitar procedimientos dentales invasivos. Durante el tratamiento con Prolia debe mantenerse una buena práctica de higiene bucal. En los pacientes que desarrollen ONM durante el tratamiento con Prolia, la cirugía dental puede empeorar su estado. Si se produce ONM durante el tratamiento con Prolia, siga el criterio clínico y elabore una pauta de tratamiento para cada paciente basada en la evaluación del riesgo/beneficio a nivel individual.
|
|
Precauciones:
Embarazo y lactancia: Embarazo: No hay datos adecuados en mujeres embarazadas. No se recomienda el uso de Prolia en mujeres embarazadas. A niveles de exposición AUC hasta 100 veces superiores al nivel de exposición en seres humanos (60 mg cada 6 seis meses), el Denosumab no mostró indicios de que afectara la fertilidad o dańara al feto en monos macacos sometidos a estudios de toxicidad en el desarrollo (véase Datos preclínicos de seguridad). Los estudios realizados en ratones genéticamente noqueados sugieren que la ausencia del RANKL podría interferir en el desarrollo de los nódulos linfáticos en el feto y deteriorar la dentición y el crecimiento óseo postnatales; también podría interferir en la maduración de las glándulas mamarias de las madres, conduciendo posparto, a una lactancia inadecuada. Lactancia: Se desconoce si el Denosumab se excreta en la leche humana. Como el Denosumab tiene el potencial de ocasionar reacciones adversas en lactantes que están amamantando, el médico tratante deberá decidir entre suspender la lactancia o suspender la administración del medicamento. Efectos en la capacidad de conducir y utilizar maquinaria: No se han realizado estudios sobre el efecto en la capacidad de conducir o utilizar maquinaria pesada en pacientes que reciben tratamiento con Denosumab.
|
|
Interacciones Medicamentosas:
No se han realizado estudios sobre interacciones medicamentosas con Prolia.
|
|
Sobredosificación:
No se dispone de datos obtenidos de estudios clínicos en relación con la sobredosificación de Prolia. El Denosumab ha sido administrado en estudios clínicos utilizando dosis de hasta 180 mg cada 4 semanas (dosis acumuladas de hasta 1.080 mg durante 6 meses).
|
|
Incompatibilidades:
Este medicamento no debe mezclarse con otros medicamentos.
|
|
Conservación:
Precauciones especiales de almacenamiento: Almacenar refrigerado (2°C - 8°C). No congelar. Conservar la jeringa prellenada en la caja externa a fin de protegerla de la luz directa. No agitar. Si se retira de refrigeración, Prolia debe conservarse a temperatura ambiente controlada (hasta 25°C (77°F)) en su caja original y deberá utilizarse en un lapso de 30 días.
|
|
Observaciones:
Vida de anaquel: La fecha de caducidad se indica en el empaque. Naturaleza y contenido del empaque: Prolia es un producto estéril y libre de conservadores. Jeringa: Jeringa prellenada de uso único con aguja de acero inoxidable de calibre 27. Tamańo de empaque de a una, presentada en un envase alveolado (jeringa prellenada con o sin protector de aguja) o en un empaque no alveolado (sólo jeringas prellenadas). La cubierta de la aguja de la jeringa prellenada contiene caucho natural seco, el cual es un derivado del látex (Véase Instrucciones para su Uso/Manejo).
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}