 |
Composición:
Comprimido de 200 mg: Cada comprimido contiene: 217 mg de Clorhidrato de Pazopanib, equivalentes a 200 mg de Pazopanib base libre. Comprimidos en forma de cápsula modificada, rosados, con las letras GS JT grabadas en uno de los lados. Lista de excipientes: Núcleo del comprimido 200 mg: Estearato de Magnesio, Celulosa Microcristalina, Povidona (K30), Glicolato de Almidón Sódico. Recubrimiento pelicular del comprimido de 200 mg (Opadry rosa): Hipromelosa, Oxido de Hierro Rojo (E172), Macrogol/PEG 400, Polisorbato 80, Dióxido de Titanio (E171).
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: Agentes antineoplásicos-inhibidor de la proteína quinasa. Código ATC: L01XE11.
|
|
Indicaciones:
Carcinoma de células renales (RCC): Votrient está indicado en el tratamiento de carcinoma de células renales en estado avanzado y/o metastásico. Sarcoma de partes blandas (SPB): Votrient está indicado para el tratamiento de pacientes con Sarcoma de Partes Blandas avanzado (SPB) que han recibido quimioterapia previa. La población del ensayo de Fase III excluyó a pacientes con tumor estromal gastrointestinal (GIST, por sus siglas en inglés) o sarcoma de partes blandas adipocítico.
|
|
Propiedades:
Mecanismo de acción: Votrient es un potente inhibidor tirosina cinasa (ITK) multi-objetivo oral de los receptores del factor de crecimiento endotelial vascular (VEGFR)-1, -2 y -3; del receptor del factor de crecimiento derivado de plaquetas (PDGFR)- a y -ß y del receptor del factor de la célula progenitora (c-KIT), con valores de IC50 de 10, 30, 47, 71, 84 y 74 nM, respectivamente. En experimentos preclínicos, pazopanib inhibió en forma dosis-dependiente, la autofosforilación inducida por ligandos de los receptores VEGFR-2, c-Kit y PDGFR-ß en las células. In vivo, pazopanib inhibió la fosforilación de VEGFR-2 inducida por VEGF en pulmones de ratones, la angiogénesis en varios modelos animales y el crecimiento de diversos xenoinjertos de tumores humanos en ratones. Farmacocinética: Absorción: Pazopanib se absorbe oralmente alcanzando su concentración máxima en una mediana de tiempo de 2.0 a 4.0 horas después de la administración. La dosificación diaria produce un aumento del ABC de 1.23 a 4 veces. No hubo un aumento consistente en el ABC y la Cmáx al aumentar la dosis de Votrient por encima de los 800 mg. La exposición sistémica a pazopanib aumenta al administrarse con alimentos. La administración de Votrient con alimentos bajos o altos en grasa resulta en un aumento de casi el doble en ABC y Cmáx. Por ende, Votrient debe administrarse por lo menos 1 hora antes ó 2 horas después de una comida (consulte la sección Posología). La administración de un comprimido triturado (molido) de pazopanib de 400 mg aumentó el AUC(0-72) en 46% y la Cmáx aproximadamente 2 veces y disminuyó el tmáx en aproximadamente 1.5 horas en comparación con la administración del comprimido entero. Estos resultados indican que la biodisponibilidad y la tasa de absorción oral del pazopanib aumentan después de la administración del comprimido triturado (molido) en relación con la administración del comprimido entero. Por lo tanto, debido a su potencial de aumento de la exposición, los comprimidos de Votrient no deben triturarse (véase Posología). Distribución: La unión de pazopanib a la proteína plasmática humana in vivo fue mayor al 99%, sin que dependiera de la concentración dentro del rango de 10-100 µg/ml. Los estudios in vitro indican que pazopanib es un sustrato de la P-glicoproteína (Pgp) y de la proteína resistente al cáncer de mama (BCRP por sus siglas en inglés). Metabolismo: Resultados de estudios in vitro demostraron que el metabolismo de pazopanib es mediado principalmente por el CYP3A4, con contribuciones menores del CYP1A2 y CYP2C8. Eliminación: Pazopanib se elimina lentamente con una vida media promedio de 30.9 horas después de la administración de la dosis recomendada de 800 mg. La eliminación se produce principalmente a través de las heces, con una eliminación renal que representa < 4% de la dosis administrada. Poblaciones especiales: Insuficiencia renal: En un análisis farmacocinético poblacional en 408 sujetos con varios tipos de cáncer, la depuración de creatinina (30-150 ml/min) no afectó la depuración de pazopanib. No se espera que la presencia de insuficiencia renal tenga alguna influencia en la exposición a pazopanib, y no es necesario ajustar la dosis en pacientes con depuración de creatinina ł 30 ml/min. Insuficiencia hepática: La mediana de Cmax y AUC(0-24) en estado estable de pazopanib en pacientes con insuficiencia hepática leve (definida como bilirrubina normal y cualquier grado de elevaciones de ALT, o como cualquier elevación de bilirrubina hasta de 1.5 veces el límite superior del rango normal, independientemente del valor de ALT), después de una dosis diaria 1 vez al día de 800 mg/día (30.9 µg/ml, rango de 12.5-47.3 y 841.8 µg.hr/ml, rango de 600.4-1.078), es similar a la mediana en pacientes sin insuficiencia hepática (49.4 µg/ml, rango de 17.1-85.7 y 888.2 µg.hr/ml, rango de 345.5-1.482) (véase Posología). La dosis máxima tolerada de Votrient (MDT) en pacientes con insuficiencia hepática moderada (definida como una elevación de bilirrubina > 1.5 a 3 veces el límite superior del rango normal, independientemente de los valores de ALT), fue de 200 mg 1 vez al día. La mediana de los valores en estado estable de Cmax (22.4 µg/ml, rango de 6.04-32.9) y AUC(0-24) (350.0 µg.hr/ml, rango de 131.8-487.7) después de la administración de 200 mg de Votrient 1 vez al día en sujetos con insuficiencia hepática moderada, fue de aproximadamente 45% y 39%, respectivamente, de la mediana correspondiente de los valores posteriores a la administración de 800 mg 1 vez al día en sujetos con función hepática normal (véase Posología). No existen suficientes datos en pacientes con insuficiencia hepática severa (bilirrubina total > 3 veces el límite superior del rango normal, independientemente de cualquier nivel de ALT); por lo tanto, no se recomienda el uso de Votrient en estos pacientes. Estudios clínicos: Carcinoma de Células Renales (RCC): Se evaluaron la seguridad y la eficacia de Votrient en el carcinoma de células renales (RCC) en un estudio multicéntrico, controlado con placebo, doble ciego y aleatorizado. Los pacientes (N = 435) con RCC localmente avanzado y/o metastásico se aleatorizaron para que recibieran Votrient 800 mg 1 vez al día o placebo. El objetivo primario del estudio fue evaluar y comparar los grupos de tratamiento para determinar la supervivencia libre de progresión (progression-free survival, PFS). El criterio de valoración secundario fue la supervivencia global (overall survival, OS). Los otros objetivos fueron evaluar la tasa de respuesta global y la duración de la respuesta. De los 435 pacientes en este estudio, 233 no habían recibido tratamiento y 202 eran pacientes en segunda línea que habían recibido una terapia previa basada en IL-2 o INF a . El estatus de desempeńo (ECOG) era similar entre los grupos de Votrient y placebo (ECOG 0:42% contra 41%, ECOG 1: 58% contra 59%). Todos los pacientes presentaban una histología a células claras o histología predominantemente a células claras. Aproximadamente la mitad de los pacientes tenía 3 ó más órganos afectados por la enfermedad, mientras que la mayoría presentaba compromiso pulmonar (74%) y/o nódulos linfáticos (54%) como ubicación metastásica de la enfermedad en el punto de partida. Una proporción similar de pacientes en cada grupo no había recibido tratamientos previos o había sido tratada previamente con citokina (53% y 47% en el grupo de Votrient, 54% y 46% en el grupo de placebo). En el subgrupo tratado previamente con citokina, la mayoría (75%) había recibido un tratamiento a base de interferón. Una proporción similar de pacientes en cada grupo había sufrido nefrectomía previa (89% y 88% en los grupos de Votrient y placebo, respectivamente) y/o radioterapia previa (22% y 15% en los grupos de Votrient y placebo, respectivamente). El análisis primario del criterio de valoración primario de PFS se basó en la evaluación de la enfermedad mediante una revisión radiológica independiente en toda la población del estudio (primera y segunda líneas).
Ver Tabla
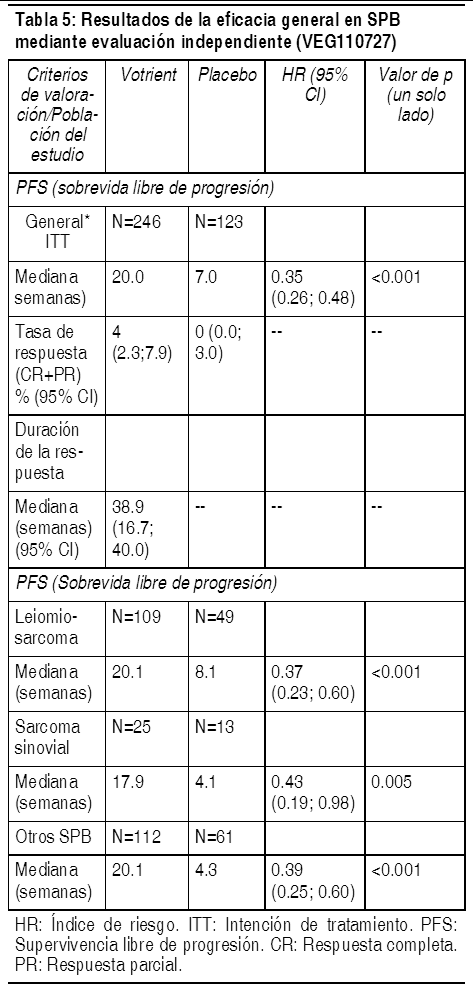
Para los pacientes que respondieron al tratamiento, la mediana de duración de la respuesta fue de 58.7 semanas, según la revisión independiente. La mediana de los resultados de sobrevida global (OS, por sus siglas en inglés) en el análisis final de sobrevida especificado en el protocolo fueron de 22.9 meses y 20.5 meses [HR = 0.91 (95% CI: 0.71; 1.16; p = 0.224)] para pacientes aleatorizados a los brazos de Votrient y placebo, respectivamente. Los resultados de OS están sujetos a un sesgo potencial, ya que el 54% de pacientes en el brazo de placebo también recibieron Votrient en la extensión de este estudio después de la progresión de la enfermedad. Sesenta y seis por ciento de los pacientes con placebo recibieron tratamiento post-estudio en comparación con el 30% de los pacientes con Votrient. En el estudio pivote, las valoraciones de la calidad de vida (quality of life, QoL) se basaron en las puntuaciones globales, ciegas, reportadas por los individuos en los 2 cuestionarios especificados por el protocolo, EORTC QLQ-C30 y EuroQoL EQ-5D. El análisis se basó en los pacientes que continuaron la terapia en ambos grupos, antes de la progresión. Las valoraciones no mostraron diferencias entre el tratamiento con Votrient o placebo (p > 0.05), lo cual indica que Votrient no produjo un efecto negativo en la calidad de vida global. En un estudio de fase 2 con 225 pacientes con carcinoma renal de células claras, localmente recurrente o metastásico, el índice de respuesta objetiva fue de 35% y la mediana de duración de la respuesta fue de 68 semanas, según la revisión independiente. Sarcoma de partes blandas (SPB): Se evaluaron la seguridad y la eficacia de Votrient en SPB en un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico. Los pacientes (N=369) con SPB avanzado que habían recibido quimioterapia previa, incluyendo tratamiento con antraciclina, o que no estaban en condiciones de recibir dicho tratamiento, fueron aleatorizados para recibir Votrient 800 mg 1 vez al día o placebo. Antes de la aleatorización, los sujetos elegibles fueron estratificados según los factores de estado de desempeńo de la OMS (WHO PS, por sus siglas en inglés) (0 ó 1) basal y por el número de líneas de tratamiento sistémico previamente recibidas para la enfermedad avanzada (0 ó 1 vs. 2+). En cada grupo de tratamiento, hubo un porcentaje ligeramente mayor de sujetos con 2+ líneas de tratamiento sistémico previo para enfermedad avanzada (58% y 55% respectivamente para los brazos de tratamiento con placebo y con Votrient) en comparación con 0 ó 1 líneas de tratamiento sistémico previo (42% y 45%, respectivamente para los brazos de tratamiento de placebo y Votrient). Hubo un poco más de sujetos con una WHO PS de 1 al inicio. La mediana de la duración del seguimiento de los sujetos (definida como la fecha de aleatorización hasta la fecha del último contacto o la muerte) fue similar para ambos brazos de tratamiento (9.36 meses para placebo [rango de 0.69 a 23.0 meses] y 10.04 meses para Votrient [rango de 0.2 a 24.3 meses]). El objetivo primario del estudio fue evaluar y comparar la sobrevida libre de progresión (PFS) entre los 2 brazos de tratamiento, con base en la población ITT, y el principal criterio secundario de valoración fue la supervivencia general (OS). El análisis inicial del objetivo primario de PFS se basó en la evaluación de la enfermedad mediante una revisión radiológica independiente en toda la población de estudio ITT.
Ver Tabla
De forma similar a las evaluaciones mediante revisión radiológica independiente, se observó una mejoría clínicamente significativa y estadísticamente significativa en la PFS basado en las evaluaciones del investigador en el brazo de Votrient en comparación con el brazo de placebo (HR: 0.39; 95% CI, 0.30 a 0.52, p <0.001). La tasa de riesgo durante el análisis interino preespecificado para la supervivencia general a favor de Votrient no fue estadísticamente significativa; la mediana de la sobrevida general en el brazo de placebo fue de 10.4 meses (IC del 95%, 8.7 a 12.7) y fue de 11.9 meses (IC del 95%, 10.7 a 15.1) en el brazo de Votrient; HR = 0,82 (IC del 97.87%: 0.59 a 1.14, p = 0.156). Información preclínica de seguridad: Carcinogénesis, mutagénesis deterioro de la fertilidad: Aunque no se han realizado estudios de carcinogenicidad definitivos con Votrient, los ratones que recibieron 1000 mg/kg/día (aproximadamente 1.5 veces la exposición clínica humana en función del ABC) durante 13 semanas presentaron lesiones proliferativas en el hígado, incluyendo focos eosinofílicos en 2 hembras y un solo caso de adenoma en otra hembra. Pazopanib no provocó dańos genéticos cuando se evaluó en los estudios de genotoxicidad (ensayo de Ames, ensayo de aberración cromosómica en linfocitos periféricos humanos y en el ensayo de micronúcleos de rata in vivo). En ratas hembras, se observó una menor fertilidad, incluyendo pérdidas pre y postimplantación y resorciones tempranas, en dosis ł 10 mg/kg/día (aproximadamente 0.2 veces la exposición clínica humana en función del ABC). La disminución del cuerpo lúteo se observó en monos que recibieron 500 mg/kg/día hasta por 34 semanas y en ratones que recibieron ł 100 mg/kg/día durante 13 semanas. Se observaron ovarios atrofiados en ratas que recibieron 300 mg/kg/día durante 26 semanas (aproximadamente igual a 0.6; 1.4 y 0.9 veces la exposición clínica humana en función del ABC, respectivamente). Pazopanib no afectó el apareamiento ni la fertilidad en ratas machos. Sin embargo, hubo reducciones en la tasa de producción de esperma, motilidad del esperma y concentraciones de esperma epididimario y testicular con dosis ł 100 mg/kg/día (aproximadamente 0.5 veces la exposición clínica humana en función del ABC) después de 15 días de dosificación. Después de 26 semanas de dosificación, hubo una disminución en los pesos testiculares y epididimario, atrofia y degeneración de los testículos con aspermia, hipospermia y cambios cribiformes en el epidídimo de ratas machos que recibieron dosis ł 30 mg/kg/día (aproximadamente 0.4 veces la exposición clínica humana en función del ABC). Pazopanib produjo efectos teratogénicos fetales (entre ellos, malformaciones cardiovasculares y osificación retardada), reducción del peso corporal fetal y mortalidad embrionaria en ratas con un nivel de dosis ł 3 mg/kg/día (aproximadamente 0.1 veces la exposición clínica humana con base en el ABC). En conejos, se observó toxicidad materna (pérdida de peso corporal, reducción del consumo de alimentos y aborto) en dosis ł 30 mg/kg/día (aproximadamente 0.007 veces la exposición clínica humana con base en el ABC), mientras que el peso fetal se redujo en dosis ł 3 mg/kg/día (consulte las secciones Embarazo y lactancia; Advertencias y precauciones). Toxicología y/o farmacología animal: En estudios de toxicología en ratas, hubo efectos en una variedad de tejidos (huesos, dientes, médula ósea, lechos ungueales, órganos reproductivos, tejidos hematológicos, rińones, glándulas suprarrenales, nódulos linfáticos, pituitaria y páncreas) consistentes con la inhibición del VEGFR y/o disrupción de las vías de seńalización del VEGF. Algunos efectos se produjeron con dosis de 3 mg/kg/día (aproximadamente 0.1 veces la exposición clínica humana con base en el ABC). Los efectos hepáticos incluyeron aumentos leves de las transaminasas hepáticas en roedores y aumentos de la bilirrubina en monos, sin una histopatología asociada a dosis que produjeran exposiciones sistémicas aproximadamente 0.1 y 0.6 veces la exposición clínica humana, respectivamente. En los estudios de toxicidad juvenil, cuando a las ratas antes del destete se les dosificó desde el día 9 post-parto hasta el día 14 post-parto, pazopanib causó mortalidades y crecimiento/maduración anormal de órganos incluyendo el rińón, pulmón, hígado y corazón, a una dosis de aproximadamente 0.1 veces la exposición clínica con base en el AUC (área bajo la curva) en adultos. Cuando las ratas posteriormente al destete fueron dosificadas desde el día 21 post-parto al día 62 post-parto, los hallazgos toxicológicos fueron similares a los de las ratas adultas a exposiciones comparables con cambios en los huesos, tráquea, dientes, glándulas adrenales, páncreas, estómago, duodeno, nódulo linfático, glándula mamaria masculina y órganos reproductivos. En las ratas, el destete ocurre al día 21 postparto, el cual equivale a la edad humana pediátrica de 2 ańos aproximadamente. Los pacientes pediátricos humanos tienen un mayor riesgo de desarrollar efectos en los huesos y dientes en comparación con los adultos, ya que estos cambios, incluyendo extremidades más cortas, estuvieron presentes en las ratas jóvenes a ł 10 mg/kg/día (igual a aproximadamente 0.1-0.2 veces la exposición clínica con base en el ABC en adultos) (véase Advertencias y Precauciones).
|
|
Posología:
La dosis recomendada de Votrient para el tratamiento de RCC o SPB es de 800 mg 1 vez al día, por vía oral. Votrient debe ingerirse sin alimentos (por lo menos 1 hora antes o 2 horas después de una comida) (consulte la sección Farmacocinética). Votrient debe ingerirse en forma completa con agua, sin triturar o fraccionar el comprimido (véase Farmacocinética). Si se omite una dosis, no debe tomarse si faltan menos de 12 horas para la dosis siguiente. Modificaciones de la dosis: Las modificaciones a las dosis, ya sea un aumento o reducción, deben hacerse en incrementos progresivos de 200 mg, según sea el caso en función de la tolerabilidad del individuo para controlar las reacciones adversas. La dosis diaria de Votrient no debe exceder los 800 mg. Poblaciones especiales de pacientes: Insuficiencia renal: No existe experiencia con el uso de Votrient en pacientes con insuficiencia renal grave en diálisis peritoneal o hemodiálisis, por lo tanto, el uso de Votrient no se recomienda en dichos pacientes. No se espera que la existencia de insuficiencia renal tenga un efecto clínicamente relevante en el perfil farmacocinético del pazopanib, dado el bajo nivel de excreción renal de pazopanib y sus metabolitos, por lo que el ajuste de la dosis no es necesario en pacientes con depuración de creatinina ł 30 ml/min (véase Eliminación). Insuficiencia hepática: Aún no se establecen completamente los perfiles de seguridad y farmacocinética del pazopanib en pacientes con insuficiencia hepática preexistente (véase Advertencias y Precauciones). No se requiere ajustar la dosis en pacientes con insuficiencia hepática leve, definida mediante los niveles de alanina aminotransferasa (ALT) y bilirrubina (véase Farmacología Clínica). La dosis de Votrient debe reducirse a 200 mg al día en pacientes con insuficiencia hepática moderada. Existen datos insuficientes en pacientes con insuficiencia hepática grave (bilirrubina total > 3 veces el límite superior del rango normal, independientemente del valor de ALT); por lo tanto, no se recomienda el uso de Votrient en estos pacientes. Nińos: No se han establecido la seguridad ni la eficacia de Votrient en nińos. (Véase Advertencias y Precauciones, Información preclínica). Personas de edad avanzada: No es necesario alterar la dosificación, frecuencia de las dosis o la vía de administración en pacientes mayores de 65 ańos.
|
|
Modo de Empleo:
No existe información relevante.
|
|
Efectos Colaterales:
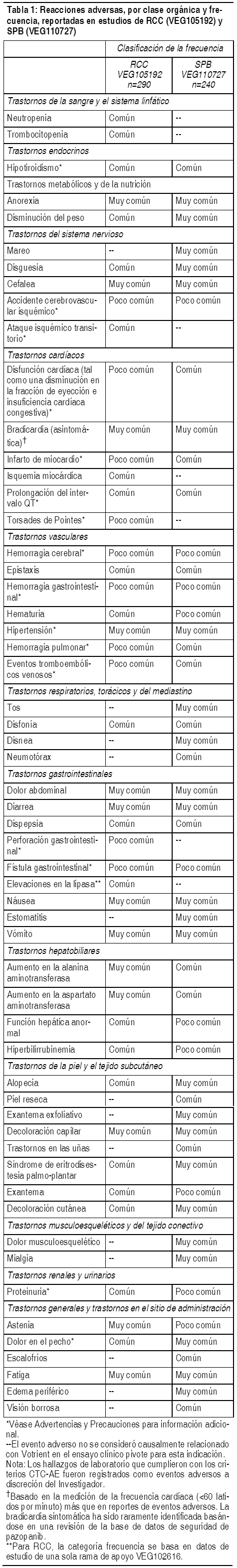
Se evaluaron la seguridad y la eficacia de Votrient en carcinoma de células renales (RCC) en un estudio multicéntrico, controlado con placebo, doble ciego y aleatorizado. Los pacientes con RCC localmente avanzado se aleatorizaron para que recibieran Votrient 800 mg 1 vez al día (N=290) o placebo (N=145). La mediana de duración del tratamiento fue de 7.4 meses para el grupo de Votrient y de 3.8 meses para el grupo de placebo. Se evaluaron la seguridad y la eficacia de Votrient en sarcoma de partes blandas en un estudio aleatorizado, doble ciego, multicéntrico, controlado con placebo. Los pacientes (N = 369) con SPB avanzado que habían recibido tratamiento previo con antraciclina, o que no estaban en condiciones de recibir dicho tratamiento, fueron aleatorizados para recibir Votrient 800 mg 1 vez al día (N = 246) o placebo (N = 123). La mediana de la duración del tratamiento fue de 4.5 meses para el brazo de Votrient y de 1.9 meses para el brazo de placebo. A continuación, se enumeran las reacciones adversas según la clasificación de órganos y sistemas corporales del MedDRA. Se utilizó la siguiente nomenclatura para la clasificación de frecuencia: Muy común ł 1 en 10; Común ł 1 en 100 y < 1 en 10; Poco común ł 1 en 1.000 y < 1 en 100; Raros ł 1 en 10.000 y < 1 en 1.000. Las categorías se asignaron en función de las frecuencias absolutas en los datos del estudio clínico.
Ver Tabla
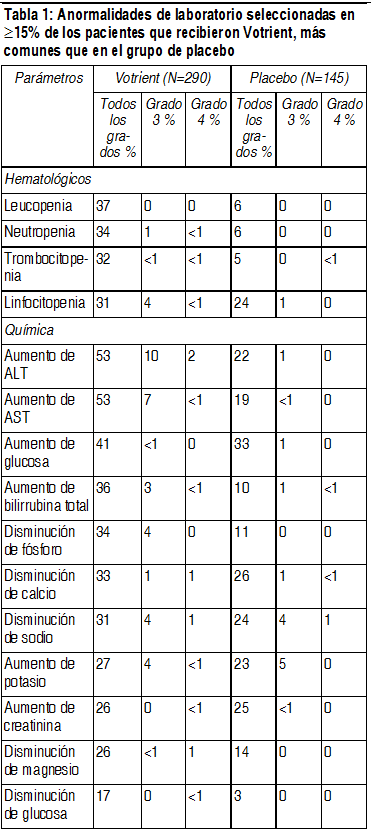
La Tabla 2 presenta anomalías de laboratorios que ocurrieron en el ł 15% de los pacientes que recibieron Votrient en los estudios pivote de RCC. Los grados se basan en NCI CTCAE.
Ver Tabla
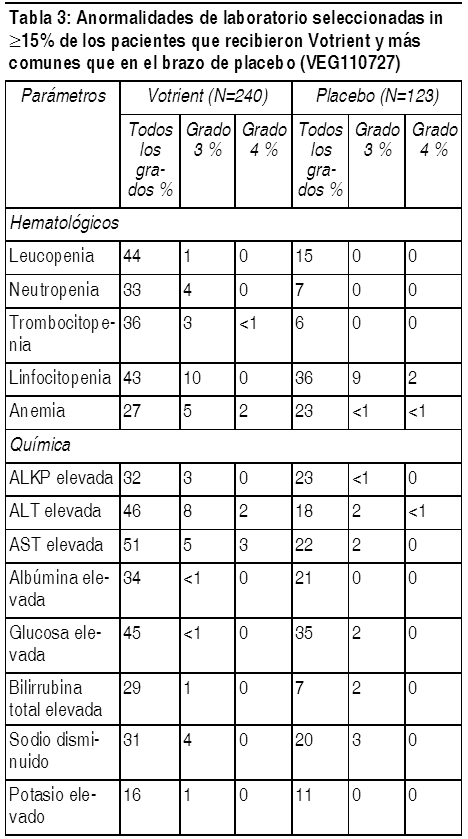
La Tabla 3 presenta las anormalidades de laboratorio que ocurren en ł 15% de los pacientes que recibieron Votrient en el estudio pivote de SPB. Los grados están basados en el NCI CTCAE.
Ver Tabla
Datos post-comercialización: Las siguientes reacciones adversas se han identificado durante el uso post-aprobación de Votrient. Esto incluye reportes de caso espontáneos así como eventos adversos serios de estudios en curso, estudios de farmacología clínica y estudios exploratorios en indicaciones no aprobadas. Infecciones e infestaciones: Poco comunes: Infecciones (con o sin neutropenia). Trastornos de sangre y sistema linfático: Raros: Microangiopatía trombótica (incluyendo púrpura trombótica trombocitopénica y síndrome urémico hemolítico). Trastornos del sistema nervioso: Raros: Síndrome de encefalopatía posterior reversible. Trastornos gastrointestinales: Poco comunes: Pancreatitis.
|
|
Contraindicaciones:
Votrient está contraindicado para pacientes con hipersensibilidad a algunos de sus componentes.
|
|
Advertencias:
Efectos hepáticos: Durante la utilización de Votrient, se han reportado casos de insuficiencia hepática (entre ellos, fatales). En los estudios clínicos con Votrient, se observaron aumentos en las transaminasas séricas (ALT, AST) y bilirrubina (consulte la sección Efectos colaterales). En la mayoría de los casos se han reportado elevaciones aisladas en los niveles de ALT y AST, sin incrementos concomitantes en los niveles de fosfatasa alcalina o bilirrubina. La gran mayoría de todas las elevaciones de transaminasas (más de 90%) de cualquier grado ocurrieron durante las primeras 18 semanas. Los grados se basan en el Criterio de Terminología Común para Eventos Adversos del Instituto Nacional del Cáncer, versión 3 (NCI CTCAE). Controle la función hepática con pruebas sanguíneas antes de comenzar el tratamiento con Votrient y por lo menos 1 vez cada 4 semanas durante por al menos los primeros 4 meses de tratamiento y además según la necesidad clínica. Luego de esta etapa, se debe realizar un control periódico. Se proporcionan las siguientes directrices para pacientes con valores iniciales (basales) de bilirrubina total Ł 1.5 veces el límite superior del rango normal y AST y ALT Ł 2 veces el límite superior del rango normal. Los pacientes con aumentos aislados de las pruebas hepáticas de entre 3 y Ł 8 veces el límite superior del rango normal pueden continuar el tratamiento con Votrient, con un control semanal de la función hepática hasta que los valores de ALT retornen a Grado 1 (NCI CTCAE) o al nivel del punto de partida. Los pacientes con aumentos de ALT > 8 veces el límite superior del rango normal deben interrumpir el tratamiento con Votrient hasta que el nivel retorne a Grado 1 (NCI CTCAE) o al nivel del punto de partida. Si se considera que el beneficio potencial de reiniciar el tratamiento con Votrient supera el riesgo de hepatotoxicidad, entonces se debe reintroducir Votrient con una dosis reducida de 400 mg 1 vez al día y medir las pruebas hepáticas sanguíneas semanalmente, durante 8 semanas (véase Posología). Después de la reintroducción de Votrient, si se vuelven a presentar elevaciones en los niveles de ALT de > 3 veces el límite superior del rango normal, entonces se deberá discontinuar permanentemente el tratamiento con pazopanib. Si se presentan elevaciones en los niveles de ALT de > 3 veces el límite superior del rango normal de manera concurrente con elevaciones en los niveles de bilirrubinemia > 2 veces el límite superior del rango normal, se deberá discontinuar permanentemente el tratamiento con Votrient. Se debe monitorear estos pacientes, hasta que los valores regresen al Grado 1 (NCI CTCAE) o inicial (basal). El pazopanib es un inhibidor de UGT1A1. Puede presentarse hiperbilirrubinemia indirecta (no conjugada) leve en pacientes con síndrome de Gilbert. En pacientes que sólo tengan una hiperbilirrubinemia indirecta leve, síndrome de Gilbert confirmado o presuntivo y elevación de ALT de > 3 veces el límite superior del rango normal, pazopanib debe administrarse según las recomendaciones establecidas para elevaciones aisladas de ALT. El uso concomitante de Votrient y simvastatina aumenta el riesgo de experimentar elevaciones de ALT (véase Interacciones), por lo cual debe realizarse con precaución y con monitoreo estrecho. Además de recomendar que los pacientes con insuficiencia hepática leve sean tratados con 800 mg de Votrient 1 vez al día, y mediante la reducción de la dosis inicial a 200 mg por día en pacientes con insuficiencia moderada, no se han establecido otros lineamientos para modificar la dosis con base en los resultados de las pruebas de función hepática en suero durante el tratamiento en pacientes con insuficiencia hepática preexistente. Hipertensión: En estudios clínicos con pazopanib, han ocurrido eventos de hipertensión incluyendo crisis hipertensiva. La presión arterial debe estar bien controlada antes de comenzar el tratamiento con Votrient. Se debe monitorear la presión arterial poco tiempo después del inicio del tratamiento (no más de 1 semana después de iniciar Votrient) y en forma frecuente para asegurar el control de la presión arterial; y tratar tempranamente con una combinación de terapia antihipertensiva estándar y reducción o suspensión de Votrient según necesidad clínica (consulte la sección Posología, Efectos colaterales). La hipertensión (presión sanguínea sistólica ł 150 o presión sanguínea diastólica ł 100 mm Hg) se produce en fase temprana durante el tratamiento con Votrient (aproximadamente el 40% de los casos ocurre antes del noveno día y aproximadamente el 90% ocurrió en las primeras 18 semanas). Votrient debe discontinuarse si hay evidencia de crisis hipertensiva o si la hipertensión es grave y persiste a pesar de la terapia antihipertensiva y la reducción de la dosis de Votrient. Síndrome encefalopatía reversible posterior (PRES por sus siglas en inglés)/Síndrome leucoencefalopatía posterior reversible (RPLS por sus siglas en inglés): Se ha reportado PRES/RPLS en asociación con Votrient. PRES/RPLS se pueden presentar con cefalea, hipertensión, convulsiones, letargia, confusión, ceguera y otras alteraciones visuales y neurológicas, y pueden ser fatales. Discontinuar permanentemente Votrient en pacientes que desarrollen PRES/RPLS. Disfunción cardíaca: En ensayos clínicos con Votrient, han ocurrido eventos de disfunción cardíaca tales como insuficiencia cardíaca congestiva y disminución en la fracción de eyección del ventrículo izquierdo (LVEF, por sus siglas en inglés). Se reportó insuficiencia cardíaca congestiva en 3 de los 240 sujetos (1%) en el estudio clínico fase III con SPB. En este estudio se detectaron disminuciones en LVEF en los sujetos que tuvieron una medición posterior al inicio del tratamiento en el 11% de los sujetos (16/142) en el brazo de Votrient en comparación con el 5% (2/40) en el brazo de placebo. Catorce de los 16 sujetos en el brazo de Votrient tenían hipertensión concurrente, la cual pudo haber empeorado la disfunción cardiaca en los pacientes en riesgo (por ej., aquéllos con tratamiento previo con antraciclina) al incrementar la postcarga cardíaca. La presión sanguínea debe monitorearse y manejarse de inmediato utilizando una combinación de tratamiento anti-hipertensivo y modificación de dosis de Votrient (interrupción y reinicio a una dosis reducida con base en el juicio clínico). Los pacientes deben ser monitoreados cuidadosamente en busca de signos o síntomas clínicos de insuficiencia cardíaca congestiva. Se recomienda una evaluación inicial y periódica de LVEF en pacientes en riesgo de disfunción cardíaca. Prolongación del intervalo QT y torsades de pointes: En estudios clínicos con Votrient, se han producido episodios de prolongación del QT o torsade de pointes (consulte la sección Efectos colaterales). Votrient debe emplearse con precaución en pacientes con antecedentes de prolongación del intervalo QT, pacientes que toman antiarrítmicos u otros medicamentos que puedan prolongar potencialmente el intervalo QT o aquellas personas con una enfermedad cardíaca preexistente relevante. Cuando se utilice Votrient, se recomienda realizar un monitoreo inicial (basal) periódico de los electrocardiogramas y mantener los electrolitos (calcio, magnesio, potasio) dentro del nivel normal. Eventos trombóticos arteriales: En estudios clínicos con Votrient, se observaron infartos de miocardio, angina, accidente cerebrovascular isquémico y ataque isquémico transitorio (consulte la sección Efectos colaterales). Se han observado eventos fatales. Votrient debe utilizarse con precaución en pacientes con un mayor riesgo de eventos trombóticos o que hayan tenido un evento trombótico dentro de los 6 meses previos. Se debe tomar una decisión con respecto al tratamiento, en función de la valoración del riesgo/beneficio de cada paciente. Eventos tromboembólicos venosos: En estudios clínicos con Votrient, han ocurrido eventos tromboembólicos venosos, incluyendo trombosis venosa y embolia pulmonar fatal. La incidencia fue mayor en la población con SPB (5%) que en la población con RCC (2%). Microangiopatía trombótica: Se ha reportado microangiopatía trombótica (TMA por sus siglas en Inglés) en estudios clínicos de Votrient usado como monoterapia, en combinación con bevacizumab, y en combinación con topotecan (véase Efectos colaterales). Discontinuar permanentemente Votrient en pacientes que estén desarrollando TMA. Se ha observado reversión de TMA después de la descontinuación del tratamiento. Votrient no está indicado para uso en combinación con otros agentes. Eventos hemorrágicos: En estudios clínicos con Votrient, se han informado eventos hemorrágicos (consulte la sección Efectos colaterales). Han sucedido eventos hemorrágicos fatales. No se ha estudiado el uso de Votrient en pacientes con antecedentes de hemoptisis, hemorragia cerebral o gastrointestinal con significación clínica en los 6 meses previos. Votrient debe emplearse con precaución en pacientes con un riesgo significativo de hemorragia. Perforaciones y fístulas gastrointestinales: En estudios clínicos con Votrient, se han producido eventos de perforaciones o fístulas gastrointestinales (GI) (consulte la sección Efectos colaterales). Se han presentado eventos de perforación gastrointestinal fatales. Votrient debe emplearse con precaución en pacientes con riesgo de perforaciones o fístulas GI. Cicatrización de heridas: No se han realizado estudios formales sobre el efecto de Votrient en la cicatrización de heridas. Dado que los inhibidores del factor de crecimiento endotelial vascular (vascular endotelial growth factor, VEGF) pueden afectar la cicatrización de heridas, el tratamiento con Votrient debe interrumpirse por lo menos 7 días antes de la fecha de las cirugías programadas. La decisión de retomar Votrient luego de la cirugía debe basarse en el juicio clínico de una cicatrización adecuada. Votrient debe discontinuarse en pacientes con dehiscencia de heridas. Hipotiroidismo: En estudios clínicos con Votrient, se han presentado eventos de hipotiroidismo (consulte la sección Efectos colaterales). Se recomienda un control proactivo de las pruebas de función tiroidea. Proteinuria: En estudios clínicos con Votrient se ha reportado proteinuria (véase Efectos colaterales). Se recomienda realizar un análisis de orina inicial (basales) y estudios periódicos durante el tratamiento .Se debe monitorear a los pacientes si la proteinuria empeora. Se debe descontinuar el uso de Votrient si el paciente desarrolla síndrome nefrótico. Infecciones: Se han reportado casos de infecciones graves (con o sin neutropenia), en algunos casos con un desenlace fatal. Combinación con otros tratamientos anticancerígenos sistémicos: Los estudios clínicos de Votrient en combinación con pemetrexed (cáncer pulmonar de células no pequeńas (NSCLC, por sus siglas en inglés) y lapatinib (cáncer cérvico-uterino) terminaron de forma temprana debido a preocupaciones con respecto al aumento de la toxicidad y/o mortalidad, y no se ha establecido una dosis de combinación segura y efectiva con estos regímenes. Votrient no está indicado para su uso en combinación con otros agentes. Toxicidad animal juvenil: Debido a que el mecanismo de acción de Votrient puede afectar gravemente el crecimiento y maduración orgánica durante el desarrollo post-natal temprano (véase Datos Preclínicos de Seguridad), Votrient no debe administrarse a pacientes humanos menores de 2 ańos de edad. Embarazo: Estudios preclínicos en animales han demostrado toxicidad reproductiva (Información preclínica sobre seguridad). Si se utiliza Votrient durante el embarazo o si la paciente queda embarazada durante el tratamiento con Votrient, se le deben explicar a la paciente los riesgos potenciales para el feto. Se debe aconsejar a las mujeres en edad reproductiva que eviten quedar embarazadas durante el tratamiento con Votrient (consulte Embarazo y lactancia).
|
|
Precauciones:
Embarazo y lactancia: Fertilidad: Votrient puede afectar la fertilidad en hombres y mujeres. En estudios de toxicidad reproductiva femenina en ratas, se observó una disminución de la fertilidad femenina (consulte la sección Información preclínica sobre la seguridad). Embarazo: No existe información adecuada sobre el uso de Votrient en mujeres embarazadas. Los estudios en animales han demostrado toxicidad reproductiva (consulte la sección Información preclínica sobre la seguridad). Se desconoce el riesgo potencial para los seres humanos. Votrient no debe utilizarse durante el embarazo, a menos que la afección clínica de la mujer requiera tratamiento con Votrient. Si se utiliza Votrient durante el embarazo o si la paciente queda embarazada durante el tratamiento con Votrient, se le deben explicar a la paciente los riesgos potenciales para el feto. Se aconseja a las mujeres en edad reproductiva que utilicen un método anticonceptivo adecuado y que eviten quedar embarazadas durante el tratamiento con Votrient. Lactancia: No se ha establecido la seguridad del uso de Votrient durante la lactancia. Se desconoce si pazopanib se excreta en la leche humana. Se debe interrumpir la lactancia durante el tratamiento con Votrient. Efectos en la habilidad para conducir automóviles y usar maquinarias: No se han realizado estudios que investiguen el efecto de Votrient en la capacidad para conducir u operar maquinarias. No se anticiparía un efecto dańino en tales actividades en función de la farmacología de pazopanib. Debe tenerse en cuenta el estado clínico del paciente y el perfil de eventos adversos de Votrient, cuando se considere la capacidad del paciente para realizar tareas que requieran de habilidades motoras, cognitivas u opinión.
|
|
Interacciones Medicamentosas:
Debe evitarse el tratamiento concomitante con inhibidores potentes de CYP3A4, glicoproteína P (P-gp) debido al riesgo de aumento de la exposición a pazopanib (véase Interacciones). Debe tenerse en cuenta la selección de productos medicinales concomitantes con mínimo o sin potencial para inhibir CYP3A4 o P- gp. Medicamentos que inhiben o inducen las enzimas 3A4 del citocromo P450: Estudios in vitro indican que el metabolismo oxidativo de pazopanib en los microsomas del hígado humano está mediado principalmente por el CYP3A4, con contribuciones menores del CYP1A2 y CYP2C8. Por lo tanto, los inhibidores e inductores del CYP3A4 pueden alterar el metabolismo de pazopanib. Inhibidores del CYP3A4, P-gp, proteínas de resistencia del cáncer de mama (BCRP por sus siglas en inglés): Pazopanib es un substrato para CYP3A4, P-gp y BCRP. La administración concomitante de pazopanib (400 mg 1 vez al día) con el inhibidor potente de CYP3A4 y P-gp, ketoconazol (400 mg 1 vez al día) durante 5 días consecutivos, resultó en un 66% y 45% de aumento del AUC(0-24) y Cmax, respectivamente, de pazopanib, en comparación con la administración de pazopanib solo (400 mg 1 vez al día por 7 días). La Cmax y el AUC de pazopanib aumentaron menos cuando se administraron aumentos proporcionales de dosis de 50 mg a 2000 mg. Por lo tanto, una dosis reducida de 400 mg pazopanib 1 vez al día en presencia de un inhibidor potente de CYP3A4 resultará, en la mayoría de los pacientes, en una exposición sistémica similar a la observada después de la administración de 800 mg de pazopanib solo 1 vez al día. Sin embargo, algunos pacientes pueden presentar una mayor exposición al pazopanib a la observada después de la administración de pazopanib solo. La coadministración de Votrient con otros inhibidores fuertes de la familia del CYP3A4 (por ejemplo, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, voriconazol) puede incrementar las concentraciones de pazopanib. El jugo de pomelo también puede aumentar las concentraciones plasmáticas de pazopanib. La administración de 1500 mg de lapatinib, un sustrato e inhibidor débil del CYP3A4, Pgp y BCRP con 800 mg de Votrient produjo un aumento aproximado de 50% a 60% en la media del ABC(0-24) y la Cmáx de pazopanib, en comparación con la administración de 800 mg de Votrient solo. La coadministración de Votrient con un inhibidor de CYP3A4, Pgp y BCRP, como el lapatinib, resultará en un aumento de las concentraciones plasmáticas de pazopanib. Debe evitarse el uso concomitante de pazopanib con un potente inhibidor de CYP3A4. Si no hay disponible una alternativa medicamente aceptable al inhibidor potente de CYP34A, debe reducirse la dosis de pazopanib a 400 mg al día durante la administración concomitante (véase Advertencias y Precauciones). Debe considerarse reducción adicional de la dosis si se observan eventos adversos posiblemente relacionados con el medicamento. Se debe evitar la combinación de inhibidores fuertes de P-gp o se debe recomendar un medicamento concomitante alternativo con un poder de inhibición mínimo o nulo de P-gp. Inductores del CYP3A4: Los inductores del CYP3A4, como rifampicina, pueden disminuir las concentraciones plasmáticas de pazopanib. Se recomienda la selección de un medicamento concomitante alternativo con un poder de inducción enzimática mínimo o nulo. Efectos de Votrient en los sustratos del CYP: Estudios in vitro con microsomas de hígado humano mostraron que pazopanib inhibía las enzimas del CYP 1A2, 3A4, 2B6, 2C8, 2C9, 2C19 y 2E1. La inducción potencial del CYP3A4 humano se demostró en un ensayo in vitro con PXR humano. Los estudios farmacológicos clínicos que utilizaron Votrient 800 mg 1 vez al día, han demostrado que Votrient no tiene un efecto con relevancia clínica en la farmacocinética de la cafeína (sustrato sonda del CYP1A2), warfarina (sustrato sonda del CYP2C9) u omeprazol (sustrato sonda del CYP2C19) en pacientes con cáncer. Votrient produjo un aumento de aproximadamente 30% en la media del ABC y la Cmáx de midazolam (sustrato sonda del CYP3A4) y aumentos de 33% a 64% en la relación de las concentraciones de dextrometorfano a dextrorfano en la orina, después de la administración oral de dextrometorfano (sustrato sonda del CYP2D6). La coadministración de Votrient 800 mg 1 vez al día y paclitaxel 80 mg/m2 (sustratos del CYP3A4 y CYP2C8) una vez por semana produjo una media de aumento de 26% y 31% en el ABC y la Cmáx de paclitaxel, respectivamente. Efectos de Votrient sobre otras enzimas y los transportadores. Estudios in vitro también han demostrado que el pazopanib es un potente inhibidor del UGT1A1 y OATP1B1 con IC50 de 1.2 y 0.79 µM, respectivamente. Votrient puede aumentar las concentraciones de fármacos eliminados principalmente mediante UGT1A1 y OATP1B1. Efecto del uso concomitante de Votrient y simvastatina: El uso concomitante de Votrient y simvastatina aumenta la incidencia de elevaciones de ALT. En los estudios de monoterapia con Votrient, se reportó ALT > 3 veces el límite superior del rango normal en 126/895 (14%) de los pacientes que no utilizaron estatinas, en comparación con 11/41 (27%) pacientes que presentaban uso concomitante de simvastatina (p = 0.038). Si un paciente que está recibiendo simvastatina concomitante desarrolla elevaciones de ALT, siga los lineamientos para la posología de Votrient y discontinúe simvastatina (véase Advertencias y Precauciones). No existen suficientes datos para evaluar el riesgo de la administración concomitante de estatinas alternativas y Votrient. Efecto de los alimentos sobre Votrient: La administración de Votrient con alimentos bajos o altos en grasa resulta en un aumento de casi el doble en el ABC y la Cmáx. Por ende, Votrient debe administrarse por lo menos 1 hora antes o 2 horas después de una comida (consulte la sección Posología).
|
|
Sobredosificación:
Se han evaluado dosis de Votrient de hasta 2000 mg. Se observaron fatiga de Grado 3 (toxicidad limitante de dosis) e hipertensión de Grado 3, cada una observada en 1 de 3 pacientes dosificados con 2000 y 1000 mg diarios, respectivamente. Síntomas y signos: Actualmente, la experiencia con sobredosis de Votrient es limitada. Tratamiento: El manejo posterior debe ser según se indique clínicamente o recomiende el centro nacional de intoxicaciones, cuando esté disponible. No se prevé que la hemodiálisis aumente la eliminación de pazopanib, ya que éste no se excreta significativamente por vía renal y está altamente unido a las proteínas plasmáticas.
|
|
Incompatibilidades:
No existen incompatibilidades conocidas.
|
|
Conservación:
Vida útil: La fecha de vencimiento se encuentra indicada en el envase. Precauciones especiales de almacenamiento: No almacenar por encima de 30şC.
|
|
Presentaciones:
Naturaleza y contenido del recipiente: Comprimidos de 200 mg: Frascos de polietileno de alta densidad (HDPE) con cierres de polipropileno a prueba de nińos.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}