 |
Composición:
Cada ml de solución contiene 100 mg de Levetiracetam. Excipientes: Citrato de Sodio; Monohidrato de Acido Cítrico; Metil parahidroxibenzoato, (Metilparabeno); Propil parahidroxibenzoato, (Propilparabeno); Glicirrizato de Amonio; Glicerol 85%; Maltitol, Licasina; Acesulfame Potásico; Sabor Uva; Agua Purificada. Forma farmacéutica: Solución clara e incolora.
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: Antiepilépticos; Otros Antiepilépticos. Código ATC: N03AX14.
|
|
Indicaciones:
Keppra® está indicado como monoterapia: en el tratamiento de las crisis convulsivas de inicio parcial, con o sin generalización secundaria; en pacientes mayores de 16 ańos con un nuevo diagnóstico de epilepsia. Keppra® está indicado como terapia concomitante: En el tratamiento de la crisis de inicio parcial con o sin generalización secundaria en adultos y nińos mayores de 4 ańos con epilepsia. En el tratamiento de las crisis mioclónicas en adultos y adolescentes mayores de 12 ańos con Epilepsia Mioclónica Juvenil. En el tratamiento de las crisis tónico-clónicas generalizadas primarias en adultos y adolescentes mayores de 12 ańos con Epilepsia Generalizada Idiopática.
|
|
Propiedades:
Farmacodinamia: Mecanismo de acción: El principio activo, levetiracetam, es un derivado de la pirrolidona (S-enantiómero de a -etil-2-oxo-1-pirrolidina acetamida), no relacionado químicamente con los principios activos de los antiepilépticos existentes. El mecanismo de acción de levetiracetam aún no se encuentra muy claro, pero al parecer su mecanismo de acción es distinto a los otros medicamentos antiepilépticos. Los experimentos in vitro e in vivo sugieren que levetiracetam no altera las características celulares básicas y la neurotransmisión normal. Estudios in-vitro muestran que el levetiracetam afecta el Ca2+ a nivel intraneuronal mediante inhibición parcial de las corrientes de Ca2+ de tipo N y mediante una reducción de la liberación de Ca2+ del espacio intraneuronal. Además, revierte parcialmente la reducción de las corrientes dependientes de GABA, y la entrada de la glicina inducida por el zinc y los ß-carbonilos. Además, un estudio in-vitro del levetiracetam muestra que éste se une a un punto específico del tejido cerebral del roedor. Este sitio específico es la proteína 2A de las vesículas sinápticas, la cual se cree que está involucrada en la fusión vesicular y la exocitosis del neurotransmisor. El levetiracetam y sus análogos relacionados muestran un orden en ranking de afinidad para unirse a la proteína 2A de las vesículas sinápticas que se correlaciona con la potencia de protección contra las crisis de epilepsia. Esto sugiere que la interacción entre el levetiracetam y la proteína 2A de la vesícula sináptica contribuye al mecanismo de acción antiepiléptico del producto. Efectos farmacodinámicos: El levetiracetam induce protección sobre las crisis convulsivas en un amplio rango de modelos animales en convulsiones generalizadas parciales y primarias sin tener un efecto pro convulsivante. El metabolito primario es inactivo. En humanos, la actividad en ambas condiciones epilepsia parcial y generalizada (descargas epilépticas/ respuesta fotoparoxismal) han confirmado el perfil de amplio espectro farmacológico de levetiracetam. Farmacocinética: Levetiracetam es un compuesto altamente soluble y permeable. Su comportamiento es lineal y con poca variabilidad intra e interindividual. No hay modificación del clearance luego de repetidas administraciones. El perfil farmacocinético independiente del tiempo de levetiracetam también fue confirmado después de una infusión de 1500 mg por vía intravenosa durante 4 días con dosificación 2 veces al día. No hay evidencia de variabilidad relevante de género, raza o ritmo circadiano. El perfil farmacocinético es comparado en voluntarios sanos y en pacientes con epilepsia. Dada su absorción completa y lineal, los niveles plasmáticos se pueden predecir mediante dosis oral expresado en mg/kg de peso corporal. Por lo tanto, no hay necesidad de monitorear los niveles plasmáticos del levetiracetam. Se ha demostrado que existe una correlación significativa entre la concentración en saliva y el nivel plasmático del levetiracetam en adultos y nińos (la relación saliva / plasma van de 1 a 1.7 en comprimidos orales y después de 4 horas de la administración de la solución oral). El perfil farmacocinético ha sido caracterizado después de la administración oral. Una sola dosis de 1500 mg de levetiracetam diluido en 100 ml de un diluyente compatible e infundido intravenosamente durante 15 minutos es bioequivalente a 1500 mg de levetiracetam por ingesta oral, tomado como 3 comprimidos de 500 mg. Se evaluó la administración intravenosa de dosis hasta de 4000 mg diluidos en 100 ml de cloruro de sodio al 0.9% infundido durante 15 minutos y en dosis hasta de 2500 mg diluidos en 100 ml de cloruro de sodio al 0.9% infundido durante 5 minutos. La farmacocinética y perfiles de seguridad no identificaron alguna inquietud de seguridad. Absorción: El levetiracetam es absorbido rápidamente luego de su administración oral. La biodisponibilidad absoluta de la forma oral es cercana al 100%. La concentración máxima a nivel plasmática (Cmáx) se alcanzan 1.3 horas luego de su administración. La concentración al estado estacionario se alcanza 2 días después de su administración 2 veces al día. El peak de concentración (Cmáx ) es de 31 a 43 µg/ml seguido de una dosis única de 1000 mg, ó 1000 mg repetido 2 veces al día. La absorción es dosis independiente y no se ve afectada por los alimentos. Distribución: No hay estudios de distribución en tejidos humanos. Ni levetiracetam ni su metabolito primario se unen significativamente a las proteínas del plasma (<10%). El volumen de distribución del levetiracetam es aproximadamente 0.5 a 0.7 l/kg, un valor cercano al volumen total de agua del cuerpo. La concentración máxima en plasma (Cmax) observada en 17 sujetos después de una sola dosis intravenosa de 1500 mg infundida durante 15 minutos fue de 51 ± 19 µg/mL (media aritmética ± desviación estándar). Metabolismo: Levetiracetam no es metabolizado extensamente en humanos. La vía metabólica (24% de la dosis) es mediante la hidrólisis enzimática del grupo acetamida. La producción del metabolito primario, ucb L057, no tiene soporte en el sistema citocromo P450 presente en el hígado. La hidrólisis del grupo acetamida es medible en varios tejidos del cuerpo incluido las células sanguíneas. El metabolito ucb LO57 es farmacológicamente inactivo. Otros dos metabolitos menores también fueron identificados. Uno se obtuvo por hidroxilación del anillo de pirrolidona (1.6% de la dosis), y el otro mediante la apertura del anillo de pirrolidona (0.9% de la dosis). Otros componentes sin identificar corresponden solo al 0.6% de la dosis. No se observó ínterconversión de ningún enantiómero del levetiracetam o su metabolito primario en la experiencia in-vivo. Los estudios in vitro han demostrado que levetiracetam y su metabolito principal no inhiben las isoformas principales del citocromo P450 hepático humano (CYP3A4,2 A6, 2C9,2C19,2D6,2E1 y 1 A2), la glucuronil transferasa (UGT1A1 y UGT1A6) y la actividad de la epóxido hidrolasa. Además, levetiracetam no afecta la glucuronidación in vitro del ácido valproico. En cultivos de hepatocitos humanos, levetiracetam tuvo poco o ningún efecto sobre el CYP1A2, SULT1E1 o UGT1A1. Levetiracetam provocó una leve inducción del CYP2B6 y del CYP3A4. Los datos de interacciones in vitro e in vivo con anticonceptivos orales, digoxina y warfarina indican que no se espera que exista una inducción enzimática significativa in vivo. Por consiguiente, es muy poco probable que Keppra interaccione con otras sustancias, o viceversa. Eliminación: La vida media plasmática en adultos fue de 7±1 hora, la que no varía con la dosis, ruta de administración o administración repetitiva. El promedio total del clearance corporal es de 0.96 ml/min/kg. La mayor vía de excreción es la urinaria, alcanzando una media del 95% de la dosis, (aproximadamente un 93% de la dosis se excreta dentro de las primeras 48 horas). La excreción vía fecal es aproximadamente un 0.3% de la dosis. La excreción urinaria acumulativa del levetiracetam ocurre en un 66% y de su metabolito primario en un 24% de la dosis, durante las primeras 48 horas. El clearance renal del levetiracetam y del ucb LO57 es de 0.6 y de 4.2 ml/min./kg respectivamente, indicando que el levetiracetam es excretado mediante filtración glomerular con la subsecuente reabsorción tubular y el metabolito primario también es excretado por secreción activa tubular junto con filtración glomerular. La eliminación del levetiracetam se correlaciona con el clearance de creatinina. Poblaciones especiales de pacientes: Nińos (4 a 12 ańos): Siguiendo la administración de una dosis oral (20 mg/kg) en nińos epilépticos (6 a 12 ańos), la vida media del levetiracetam fue de 6 horas. El clearence ajustado al peso corporal fue aproximadamente de un 30% mayor en nińos que en adultos. Tras la administración de repetidas dosis orales (20 a 60 mg/kg/día) a nińos epilépticos (4 a 12 ańos), el levetiracetam fue absorbido rápidamente. El peak de la concentración en el plasma se observó entre 0.5 a 1 hora después de la administración de la dosis. Se observó un comportamiento lineal de la droga. La vida media es de aproximadamente 5 horas. El clearance corporal aparente fue de 1.1 ml/min/kg. En el análisis farmacocinético de la población realizado en pacientes de 1 mes a 16 ańos de edad, el peso corporal tuvo una correlación significativa con la depuración aparente (la depuración aumentó con el aumento en el peso corporal) y volumen aparente de distribución. La edad también tuvo una influencia sobre ambos parámetros. Este efecto fue pronunciado en los lactantes más jóvenes, y disminuyó a medida que la edad aumentó, y se volvió insignificante alrededor de los 4 ańos de edad. En ambos análisis farmacocinéticos poblacionales, hubo un aumento de cerca del 20% de la depuración aparente de levetiracetam cuando se co-administró con un AED inductor de enzimas. Ancianos: En ancianos, la vida media del producto aumenta en un 40% (10 a 11 horas). Esto está relacionado con la disminución de la función renal en esta población. Insuficiencia renal: El clearance renal aparente corporal de ambos, el levetiracetam y su metabolito primario se correlaciona con el clearance de creatinina. Por esto se recomienda ajustar la dosis diaria de mantenimiento de Keppra, basado en el clearance de creatinina en pacientes con insuficiencia renal moderada a severa. En la fase terminal de falla renal, en sujetos anúricos, la vida media del fármaco fue aproximadamente de 25 a 3.1 horas en períodos interdiálisis e intradiálisis, respectivamente. La fracción que se removió del levetiracetam durante una sesión de diálisis de 4 horas fue de 51%. Insuficiencia hepática: En sujetos con insuficiencia hepática leve a moderada, no hubo una modificación relevante del clearance de levetiracetam. En la mayoría de los sujetos con insuficiencia hepática severa el clearance de levetiracetam se redujo en más de un 50% debido a la insuficiencia renal concomitante. Estudios clínicos: No relevantes para este producto. Información no clínica: Los datos no clínicos no muestran riesgos especiales para los seres humanos según estudios convencionales de farmacología de seguridad, genotoxicidad o potencial carcinogénico. Efectos adversos no observados en los estudios clínicos, pero si vistos en la rata y en menor medida en ratones a nivel de exposición similares a los niveles de exposición humanos, y con posible repercusión en el uso clínico, fueron modificaciones hepáticas, que indican una respuesta adaptativa como por ejemplo la hipertrofia centrolobular, infiltración grasa y aumento de las enzimas hepáticas en el plasma. No se observaron efectos adversos sobre la fertilidad masculina o femenina o el rendimiento reproductivo en ratas a dosis de hasta 1800 mg/kg/día (x 6 la MRHD en base a mg/m2 o exposición) en los padres y la generación F1. Se realizaron 2 estudios de desarrollo embriofetal (EFD) en ratas con dosis de 400, 1200 y 3600 mg/kg/día. Con 3600 mg/kg/día, en sólo uno de los 2 estudios de EFD, se observó una ligera disminución en el peso fetal asociada con un incremento marginal en las variaciones esqueléticas/anomalías menores. No se observó efecto alguno en la mortalidad embrionaria, ni aumento en la incidencia de malformaciones. El NOAEL (Nivel en el que No se Observan Efectos Adversos) fue de 3600 mg/kg/día en ratas preńadas (x 12 la MRHD en mg/m2) y de 1200 mg/kg/día en fetos. Se realizaron 4 estudios de desarrollo embrio-fetal en conejos, con dosis de 200, 600, 800, 1200 y 1800 mg/kg/día. El nivel de dosis de 1800 mg/kg/día indujo una toxicidad materna marcada y una disminución del peso fetal asociada con un aumento en la incidencia de fetos con anomalías cardiovasculares/esqueléticas. El NOAEL fue <200 mg/kg/día en madres y de 200 mg/kg/día en fetos (igual a la MRHD en mg/m2). Un estudio de desarrollo perinatal y postnatal fue realizado en ratas, con dosis de levetiracetam de 70, 350 y 1800 mg/kg/día. El NOAEL fue ł 1800 mg/kg/día en hembras F0, y para supervivencia, crecimiento y desarrollo de las crías F1 hasta el destete (x 6 la MRHD en mg/m2). En animales neonatos y juveniles de ratas y perros se demostró que no hubo efectos adversos en ninguna de las etapas del desarrollo o maduración con dosis de hasta 1800 mg/kg/día (x 6 - 17 la MRHD en mg/m2).
|
|
Posología:
Vía de administración: Oral. Se puede iniciar la terapia con levetiracetam con administración intravenosa u oral. La conversión desde o hasta la administración intravenosa puede realizarse en forma directa, sin titulación de dosis. La dosis diaria total y frecuencia de administración deberán mantenerse. La solución oral puede ser diluida en un vaso de agua y puede ser administrada con o sin alimentos. La dosis diaria se administra en dos dosis divididas equitativamente. Adultos: Monoterapia: Adultos y adolescentes desde 16 ańos de edad: La dosis inicial recomendada es 250 mg 2 veces al día, la cual debe incrementarse hasta la dosis terapéutica inicial de 500 mg 2 veces al día tras 2 semanas de tratamiento. La dosis puede incrementarse en 250 mg 2 veces al día cada 2 semanas, dependiendo de la respuesta al tratamiento. La dosis máxima corresponde a 1500 mg 2 veces al día. Terapia concomitante: Adultos ( ł 18 ańos) y adolescentes (12 a 17 ańos) con un peso de 50 kg o superior: La dosis terapéutica inicial es de 500 mg 2 veces al día. Esta dosis se puede instaurar desde el primer día de tratamiento. Dependiendo de la respuesta clínica y de la tolerabilidad, la dosis diaria se puede incrementar hasta 1500 mg 2 veces al día. La modificación de la dosis se puede realizar con aumentos o reducciones de 500 mg 2 veces al día cada 2 a 4 semanas. Nińos: El médico debe prescribir la forma farmacéutica y concentración más apropiada de acuerdo con el peso y la dosis. La seguridad y eficacia del concentrado de levetiracetam para solución para infusión en lactantes y nińos menores de 4 ańos de edad han no han sido establecidas. Monoterapia: No se ha establecido la seguridad y eficacia como monoterapia en nińos y adolescentes menores de 16 ańos. No hay datos disponibles. Terapia complementaria en nińos de 4 a 11 ańos de edad y adolescentes (de 12 a 17 ańos) con un peso inferior a 50 kg: Levetiracetam solución oral es la formulación preferida para uso en nińos menores de 6 ańos. La dosis terapéutica inicial es de 10 mg/kg 2 veces al día. En función de la respuesta clínica y de la tolerabilidad, se puede aumentar la dosis hasta los 30 mg/kg 2 veces al día. Los cambios de dosis no deberían exceder de aumentos/reducciones de 10 mg/kg 2 veces al día cada 2 semanas. Se debe utilizar la menor dosis eficaz. La dosificación en nińos con un peso de 50 kg o superior es la misma que en adultos. El médico debe prescribir la forma farmacéutica y concentración más apropiada de acuerdo con el peso y la dosis. Dosificación recomendada para nińos y adolescentes:Peso: 15 kg(1). Dosis inicial: 10 mg/kg 2 veces al día: 150 mg (1.5 ml) 2 veces al día: Dosis máxima: 30 mg/kg 2 veces al día: 450 mg (4.5 ml) 2 veces al día. Peso: 20 kg(1). Dosis inicial: 10 mg/kg 2 veces al día: 200 mg (2 ml) 2 veces al día. Dosis máxima: 30 mg/kg 2 veces al día: 600 mg (6 ml) 2 veces al día. Peso: 25 kg. Dosis inicial: 10 mg/kg 2 veces al día: 250 mg dos veces al día. Dosis máxima: 30 mg/kg 2 veces al día: 750 mg 2 veces al día. Peso: A partir de 50 kg(2). Dosis inicial: 10 mg/kg 2 veces al día: 500 mg 2 veces al día. Dosis máxima: 30 mg/kg 2 veces al día: 1500 mg 2 veces al día. (1)Nińos con un peso de 25 kg o inferior deberían preferiblemente iniciar el tratamiento con Keppra 100 mg/ml solución oral. (2)La dosificación en nińos y adolescentes con un peso de 50 kg o superior es la misma que en adultos. Uso en nińos menores de 4 ańos: Keppra no está recomendado para uso en nińos menores de 4 ańos debido a la escasez de datos sobre seguridad y eficacia. Ancianos: Se recomienda ajustar la dosis en pacientes ancianos con función renal comprometida. Insuficiencia renal: La dosis diaria debe ser individual dependiendo de la función renal (véase Advertencias). Para pacientes adultos, referirse a la siguiente tabla y ajustar la dosis indicada. Para usar esta tabla de dosificación, es necesario un estimativo del clearance de creatinina del paciente en ml/min (CLcr). El CLcr en ml/min puede ser estimado a partir de la creatinina presente en el suero (mg/dl), para adultos y adolescentes que pesan 50 kg o más, a partir de la siguiente fórmula:
Ver Tabla
Entonces se ajusta el CLcr para el área de la superficie corporal (ASC) como sigue:
Ver Tabla
Ajuste de dosis para pacientes adultos y adolescentes que pesan más de 50 kg con función renal alterada:
Ver Tabla
Para nińos con insuficiencia renal, la dosis de levetiracetam necesita ser ajustada en base a la función renal, ya que la depuración de levetiracetam está relacionada con la función renal. Esta recomendación está basada en un estudio en pacientes adultos con insuficiencia renal. El CLcr en ml/min/1.73 m2 puede estimarse a partir de la determinación de creatinina sérica (mg/dl) utilizando la siguiente fórmula para adolescentes jóvenes y nińos (fórmula Schwartz):
Ver Tabla
Ver Tabla
Insuficiencia hepática: No es necesario un ajuste de dosis en pacientes con insuficiencia hepática leve a moderada. En pacientes con insuficiencia hepática severa, el clearance de creatinina puede subestimar la insuficiencia renal, por lo tanto, una reducción de 50% de la dosis diaria de mantenimiento se recomienda cuando el clearance de creatinina es < 60 ml/min/1.73 m2.
|
|
Efectos Colaterales:
Datos de ensayos clínicos y datos post-comercialización: Resumen del perfil de seguridad: El perfil de eventos adversos presentado a continuación está basado en el análisis de estudios clínicos comparativos con placebo acumulados con todas las indicaciones estudiadas, con un total de 3416 pacientes tratados con levetiracetam. Estos datos son complementados con el uso de levetiracetam en estudios de extensión sin anonimato correspondientes, así como con la experiencia post-comercialización. Las reacciones adversas más frecuentemente reportadas fueron nasofaringitis, somnolencia, cefalea, fatiga y mareo. El perfil de seguridad de levetiracetam es generalmente similar entre los grupos de edad (pacientes adultos y pediátricos) y entre las indicaciones para epilepsia aprobadas. Las reacciones adversas están ordenadas bajo los encabezados de frecuencia usando la siguiente convención: Muy común: ł 1/10. Común: ł 1/100 a <1/10. Poco común: ł 1/1000 a <1/100. Rara: ł 1/10000 a <1/1000. Muy rara: <1/10000. Desconocida (no se puede estimar a partir de los datos disponibles). Infecciones e infestaciones: Muy común: Nasofaringitis. Rara: Infección. Trastornos de la sangre y del sistema linfático: Poco común: Trombocitopenia(1), leucopenia. Rara: Pancitopenia (1,2), neutropenia.(1) Trastornos del metabolismo y de la nutrición: Común: Anorexia. Poco común: Peso disminuido (1), aumento de peso. Trastornos psiquiátricos: Común: Depresión, hostilidad/agresión, ansiedad(1), insomnio, nerviosismo/irritabilidad. Poco común: Intento de suicidio(1), ideación suicida(1), trastorno psicótico(1), comportamiento anormal(1), alucinación(1) reacción de ira(1), estado confusional(1), labilidad afectiva/cambios del estado de ánimo, agitación. Rara: suicidio consumado(1), trastorno de la personalidad, pensamiento anormal. Trastornos del sistema nervioso: Muy común: Somnolencia, cefalea. Común: Convulsión, trastorno del equilibrio, mareo, letargo, temblor. Poco común: Amnesia, alteración de la memoria, coordinación anormal/ataxia, parestesia(1), alteración de la atención. Rara: Coreoatetosis(1), discinesia(1), hiperquinesia. Trastornos oculares: Poco común: Diplopía, visión borrosa. Trastornos del oído y del laberinto: Común: Vértigo. Trastornos respiratorios, torácicos y mediastínicos: Común: Tos. Trastornos gastrointestinales: Común: Dolor abdominal, diarrea, dispepsia, vómito, náusea. Rara: Pancreatitis(1). Trastornos hepatobiliares: Poco común: Prueba anormal de función hepática(1). Rara: Insuficiencia hepática(1), hepatitis(1). Trastornos de la piel y del tejido subcutáneo: Común: Erupción. Poco común: Alopecia(1), eczema, prurito. Rara: Necrolisis epidérmica tóxica(1), Síndrome de Stevens-Johnson(1), eritema multiforme(1). Trastornos musculoesqueléticos y del tejido conjuntivo: Poco común: Debilidad muscular, mialgia. Trastornos generales y alteraciones en el lugar de administración: Común: Astenia, fatiga. Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos: Poco común: Lesión. (1)Reacciones adversas adicionadas durante la experiencia post-comercialización. (2)Supresión de médula ósea identificada en algunos de los casos. Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos. Descripción de reacciones adversas seleccionadas: El riesgo de anorexia es mayor cuando topiramato es co-administrado con levetiracetam. En algunos casos de alopecia, se observó la recuperación cuando se descontinuó levetiracetam. Población pediátrica: En pacientes de 4-16 ańos de edad, un total de 645 pacientes han sido tratados con levetiracetam en los ensayos controlados con placebo y de extensión abierta. 233 de estos pacientes fueron tratados con levetiracetam en los ensayos controlados con placebo. En ambos rangos de edad pediátricos, estos datos se complementan con la experiencia post-comercialización del uso de levetiracetam. El perfil de seguridad de levetiracetam es, en general, similar en todos los grupos de edad y en todas las indicaciones aprobadas en epilepsia. Los resultados de seguridad de los ensayos clínicos controlados con placebo en pacientes pediátricos coincidieron con el perfil de seguridad de levetiracetam en adultos excepto por las reacciones adversas psiquiátricas y de comportamiento, las cuales fueron más frecuentes en nińos que en adultos. En nińos y adolescentes de 4 a 16 ańos de edad, vómitos (muy comunes, 11.2%), agitación (común, 3.4%), cambios de humor (común, 2.1%), inestabilidad emocional (común, 1.7%), agresividad (común, 8.2%), comportamiento anormal (común, 5.6%) y letargo (común, 3.9%) fueron notificados más frecuentemente que otros rangos de edad o que en el perfil de seguridad global. En lactantes y nińos de 1 mes a menos de 4 ańos de edad, irritabilidad (muy común, 11.7%) y coordinación anormal (común, 3.3%) fueron notificados más frecuentemente que en otros grupos de edad o que en el perfil de seguridad global. Un estudio de seguridad pediátrica doble ciego controlado con placebo con diseńo de no-inferioridad ha evaluado los efectos cognitivos y neuropsicológicos de Keppra en nińos de 4 a 16 ańos de edad con crisis de inicio parcial. Se concluyó que Keppra no era diferente (no era inferior) a placebo con respecto al cambio en la puntación en la escala "Leiter-R Attention and Memory, Memory Screen Composite" desde el inicio en la población por protocolo. Los resultados relacionados con la función emocional y el comportamiento, medidos de forma estandarizada y sistemática usando un instrumento validado (cuestionario CBCL de Achenbach), indicaron un empeoramiento del comportamiento agresivo en los pacientes tratados con Keppra. Sin embargo, los sujetos que tomaron Keppra en el ensayo de seguimiento a largo plazo, abierto, no experimentaron un empeoramiento, en promedio, en su función emocional y comportamiento; en concreto, las medidas del comportamiento agresivo no empeoraron con respecto al inicio.
|
|
Contraindicaciones:
Reacciones de hipersensibilidad a la sustancia activa o a otros derivados de las pirrolidonas o a alguno de sus excipientes.
|
|
Advertencias:
Discontinuación: De acuerdo con la práctica clínica, si levetiracetam es discontinuado se recomienda disminuirlo gradualmente. (Por ejemplo, en adultos y adolescentes que pesen más de 50 kg: reducciones de 500 mg 2 veces al día cada 2 a 4 semanas; en los nińos y adolescentes que pesen menos de 50 kg: la disminución de la dosis no debe exceder los 10 mg/kg 2 veces al día cada 2 semanas). Insuficiencia renal o hepática: La administración de Keppra a pacientes con insuficiencia renal pueden requerir ajuste en sus dosis. En pacientes con función hepática severamente deteriorada, el cálculo de la función renal es recomendado antes de la selección de dosis (Véase Posología). Depresión y/o ideación suicida: Se han notificado casos de suicidio, intento de suicidio y pensamientos y comportamientos suicidas en pacientes tratados con fármacos antiepilépticos (incluyendo levetiracetam). Un metanálisis de ensayos controlados con placebo, aleatorizados, con fármacos antiepilépticos ha mostrado un pequeńo aumento del riesgo de pensamientos y comportamientos suicidas. Se desconoce el mecanismo de este riesgo. Por tanto, los pacientes deben ser monitorizados para detectar signos de depresión y/o pensamientos o comportamientos suicidas y debe considerarse el tratamiento adecuado. Se debe aconsejar a los pacientes (y a sus cuidadores) que consulten con su médico si aparecen signos de depresión y/o pensamientos suicidas. Población pediátrica: Los datos disponibles en nińos no sugieren efecto en el crecimiento ni en la pubertad. No obstante, siguen sin conocerse los efectos a largo plazo sobre el aprendizaje, inteligencia, crecimiento, función endócrina, pubertad y fertilidad en nińos. Excipientes: La solución oral de Keppra 100 mg/ml, contiene metilparahidroxibenzoato (E218) y propilparahidroxibenzoato (E216) los cuales pueden causar reacciones alérgicas (posiblemente retardadas). Esto también incluye al maltitol; pacientes con intolerancia hereditaria a la fructosa no deben consumir este medicamento. Contiene glicerol, el cual puede causar dolor de cabeza, malestar estomacal y diarrea.
|
|
Precauciones:
Embarazo y lactancia: Fertilidad: No se detectó un impacto sobre la fertilidad en estudios con animales. No hay datos clínicos disponibles, el riesgo potencial para los humanos se desconoce. Embarazo: Levetiracetam no se recomienda durante el embarazo y en mujeres en edad reproductiva que puedan tener hijos que no utilicen anticonceptivos a menos que sea claramente necesario. No hay datos suficientes sobre el uso de Keppra en mujeres embarazadas. Estudios en animales han mostrado toxicidad reproductiva. El riesgo potencial en humanos es desconocido. Keppra no debería ser usado durante el embarazo a menos que sea estrictamente necesario. Al igual que con otros medicamentos antiepilépticos, los cambios fisiológicos durante el embarazo pueden afectar a las concentraciones de levetiracetam. Se ha observado la disminución de las concentraciones plasmáticas de levetiracetam durante el embarazo. Esta disminución es más pronunciada durante el tercer trimestre (hasta el 60% de la concentración inicial antes del embarazo). Debe asegurarse un control clínico adecuado de la mujer embarazada tratada con levetiracetam. La discontinuación del tratamiento antiepiléptico puede resultar en una exacerbación de la enfermedad lo que puede resultar perjudicial para la madre y el feto. Lactancia: El Levetiracetam es excretado en la leche materna. Por lo tanto, el amamantamiento no es recomendable si la madre está recibiendo levetiracetam. Sin embargo, si durante el período de lactancia es necesario el tratamiento con levetiracetam, debe considerarse la relación beneficio/riesgo del tratamiento teniéndose en cuenta la importancia de la lactancia natural. Capacidad de realizar tareas que requieren de habilidades de juicio, motoras o cognitivas: No se han efectuado estudios sobre los efectos en la habilidad para manejar y usar máquinas. Dada la sensibilidad individual, algunos pacientes pueden experimentar somnolencia u otros síntomas relacionados con el sistema nervioso central, especialmente al inicio del tratamiento o seguido del aumento de dosis. Por ende, se debe tener precaución en aquellos pacientes que realizan trabajos demandantes, como por ejemplo personas que manejan vehículos u operan maquinarias. Se recomienda a los pacientes tratados no deben operar con máquinas hasta que se establezca que su habilidad con ellas no se ve afectada.
|
|
Interacciones Medicamentosas:
Medicamentos antiepilépticos: Datos pre-marketing de estudios clínicos indican que levetiracetam no influye en las concentraciones séricas de otros fármacos antiepilépticos como fenitoína, carbamazepina, ácido valproico, fenobarbital, lamotrigina, gabapentina y primidona; y que estos medicamentos antiepilépticos no influyen en la farmacocinética de Keppra. Como en los adultos, no hay evidencia clínica significativa de las interacciones con otros productos en pacientes pediátricos que reciben hasta 60 mg/kg/día de levetiracetam. Un estudio retrospectivo de las interacciones farmacocinéticas en nińos y adolescentes con epilepsia (4 a 17 ańos) confirma que la terapia combinada con levetiracetam administrado por vía oral, no influencia las concentraciones séricas en estado de equilibrio de carbamazepina y valproato. Sin embargo, los datos sugieren un aumento de un 20% en el clearance de levetiracetam en nińos que toman fármacos antiepilépticos que sean inductores enzimáticos. El ajuste de la dosis no es requerida. Probenecid: Con probenecid (500 mg 4 veces al día), un agente bloqueador de la secreción tubular renal, se ha demostrado que inhibe el clearance renal del metabolito primario pero no del levetiracetam. Sin embargo, la concentración de este metabolito permanece baja. Es esperable que otros productos medicinales que son excretados por secreción activa tubular también pudieran disminuir el clearance renal del metabolito. El efecto del levetiracetam sobre el probenecid no ha sido estudiado y el efecto del levetiracetam en otros productos secretados activamente como por ejemplo AINES, sulfonamidas y metotrexato, son desconocidos. Anticonceptivos orales, digoxina y warfarina: Dosis diarias de 1000 mg de levetiracetam no influencian la farmacocinética de anticonceptivos orales, (etinilestradiol y levonorgestrel); los parámetros endocrinos (hormona luteinizante y progesterona) no fueron modificados. Dosis diarias de 2000 mg de levetiracetam no influencia la farmacocinética de la digoxina y la warfarina; el tiempo de protrombina no fue modificado. La co-administración con digoxina, anticonceptivos orales y warfarina no influencian la farmacocinética del levetiracetam. Antiácidos: No hay datos sobre la influencia que ejercen los antiácidos sobre la absorción del levetiracetam. Alimentos y alcohol: La absorción total del levetiracetam no fue reducida con el consumo de alimentos, pero ésta se hace más lenta. No hay datos disponibles sobre la interacción del levetiracetam con el alcohol.
|
|
Sobredosificación:
Síntomas y signos: Somnolencia, agitación, agresión, disminución del nivel de conciencia, depresión respiratoria y coma fueron observados con la sobredosis por levetiracetam. Tratamiento: Luego de una sobredosis aguda, el estómago debe ser vaciado mediante un lavado gástrico o por la inducción del vómito. No existe antídoto específico para el levetiracetam. El tratamiento de la sobredosis será sintomático y puede incluir hemodiálisis. La eficiencia de la extracción mediante diálisis es del 60% para el levetiracetam y de un 74% para el metabolito primario.
|
|
Incompatibilidades:
No aplicable.
|
|
Conservación:
Producto terminado cerrado: de acuerdo a lo indicado en el envase. Después de abierto el envase: 2 meses almacenado a no más de 25şC.
|
|
Observaciones:
Naturaleza y contenido del envase: Botella ámbar de vidrio de 300 ml (tipo III) con tapa de polipropileno blanca de seguridad. El envase de cartón contiene una jeringa de uso oral graduada de polietileno/poliestireno) y un folleto de información al paciente.
|
|



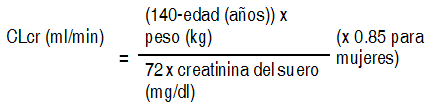{kind=link}
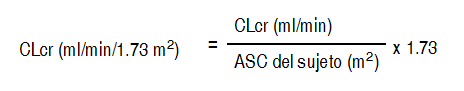{kind=link}
{kind=link}
{kind=link}
{kind=link}