 |
Composición:
Cada ml de solución para inyección intravítrea contiene: 40 mg de Aflibercept. Cada jeringa precargada contiene: 1 volumen de llenado de 165 microlitros de solución para inyección intravítrea que proporciona aproximadamente 90 microlitros de volumen extraíble. Cada jeringa precargada de dosis única proporciona una cantidad utilizable para dar una dosis única de 50 microlitros que contiene: 2 mg de aflibercept. Cada vial contiene: 1 volumen de llenado de 278 microlitros de solución para inyección intravítrea que proporciona aproximadamente 100 microlitros de volumen extraíble. Cada vial proporciona una cantidad utilizable para dar una dosis única de 50 microlitros que contiene: 2 mg de Aflibercept. Este medicamento contiene menos de 1 mmol de Sodio (23 mg) por dosis, es decir, esencialmente "exento de Sodio". Excipientes: Polisorbato 20, Fosfato Monobásico de Sodio Monohidrato, Fosfato Dibásico de Sodio Heptahidrato, Cloruro de Sodio, Sacarosa, Agua para Inyectables c.s.
|
|
Acción Terapéutica:
Forma farmacéutica: Solución para inyección intravítrea. Solución estéril, transparente, incolora a amarilla pálido, isoosmótica, acuosa de pH 6.2. Grupo farmacoterapéutico: Oftalmológicos. Agentes antineovascularización. Código ATC: S01LA05.
|
|
Indicaciones:
Eyla está indicado para el tratamiento de: La degeneración macular asociada a la edad (AMD húmeda) neovascular (húmeda). Edema macular posterior a la oclusión de la vena central de la retina (OVCR).
|
|
Propiedades:
Propiedades farmacodinámicas: Aflibercept es una proteína de fusión recombinante compuesta de porciones de dominios extracelulares del receptor 1 y 2 del VEGF humano que se fusionan al fragmento Fc de la IgG1 humana. Aflibercept es producido en células K1 de ovario de hámster chino (CHO) por tecnología de ADN recombinante. Mecanismo de acción: El factor de crecimiento endotelial vascular A (VEGF-A) y el factor de crecimiento placentario (PlGF) son miembros de la familia del VEGF de factores angiogénicos que pueden actuar como potentes factores mitogénicos, quimiotácticos y de permeabilidad vascular para las células endoteliales. El VEGF actúa por vía de 2 receptores de las tirosinquinasas, VEGFR-1 y VEGFR-2, presentes en la superficie de las células endoteliales. El PlGF se une sólo a VEGFR-1, que también está presente en la superficie de los leucocitos. La excesiva activación de estos receptores por VEGF-A puede ocasionar neovascularización patológica y excesiva permeabilidad vascular. El PlGF puede sinergizar con el VEGF-A en estos procesos y también es conocido por promover la infiltración de leucocitos y la inflamación vascular. Diversas enfermedades oculares están asociadas a neovascularización patológica, exudación vascular y/o pueden ocasionar engrosamiento y edema de la retina, que se cree que contribuye a pérdida de la visión. Aflibercept actúa como receptor señuelo soluble que se une al VEGF-A y PlGF con mayor afinidad que sus receptores naturales y, en consecuencia, puede inhibir la unión y activación de estos receptores análogos del VEGF. La constante de equilibrio de disociación (KD) de la unión de aflibercept al VEGF-A165 humano es 0.5 pM y al VEGF-A121 humano es 0.36 pM. La KD de unión al PlGF-2 humano es 39 pM. En estudios en animales, aflibercept puede prevenir la neovascularización patológica y la exudación vascular en diversos modelos diferentes de enfermedad ocular. Por ejemplo, la administración intravítrea de aflibercept a monos previno el desarrollo de neovascularización coroidea (NVC) significativa después de lesión por láser y revirtió la exudación vascular por lesiones NVC establecidas. Efectos farmacodinámicos: Degeneración macular asociada a la edad (AMD húmeda) neovascular (húmeda): La AMD húmeda se caracteriza por neovascularización coroidal (NVC) patológica. La exudación de sangre y líquido de la NVC puede causar edema retiniano y/o hemorragia subrretiniana/intrarretiniana, ocasionando pérdida de la agudeza visual. En pacientes tratados con Eylia (1 inyección mensual durante 3 meses consecutivos, seguida de 1 inyección cada mes o cada 2 meses) disminuyó el engrosamiento retiniano poco después de la iniciación del tratamiento y se redujo el tamaño lesional medio de la NVC, consistente con los resultados observados con ranibizumab 0.5 mg cada mes. En el segundo año de los estudios, los pacientes siguieron recibiendo las concentraciones de dosis a las que fueron inicialmente asignados en forma aleatoria, pero con un cronograma de dosis modificado guiado por una evaluación de resultados visuales y anatómicos con un intervalo de dosis máximo de 12 semanas, definido por el protocolo. En el estudio VIEW1 hubo disminuciones medias del grosor de la retina por tomografía de coherencia óptica (TCO) (-123, -121, -130 y -129 micrones en la semana 52 para los grupos estudiados de Eylia 0.5 mg mensual, 2 mg mensual, 2 mg cada 2 meses y ranibizumab 0.5 mg cada mes, respectivamente). También en el intervalo de 52 semanas, en el estudio VIEW2 hubo disminuciones medias del grosor de la retina por TCO (-130, -157, -149 y -139 micrones para los grupos estudiados de Eylia 0.5 mg al mes, 2 mg al mes, 2 mg cada dos meses y ranibizumab 0.5 mg cada mes, respectivamente). La reducción del tamaño NVC y la reducción en el engrosamiento retiniano en general se mantuvieron en el segundo año de los estudios. Edema macular posterior a la oclusión de la vena central de la retina (OVCR): En la OVCR, la isquemia retinal tiene lugar, e indica la liberación del VEGF que a su vez desestabiliza las uniones estrechas y promueve la proliferación de las células endoteliales. El aumento del VEGF está relacionado con la descomposición de la barrera hemato-retiniana, y esta permeabilidad vascular aumentada resulta en edema retinal, estimulación del crecimiento de las células endoteliales y neovascularización. En los pacientes tratados con Eylia (1 inyección cada mes durante seis meses) hubo una respuesta coherente, rápida y robusta en la morfología (engrosamiento retinal central [ERC] como lo evaluó la TCO). Las mejoras en la ERC media se mantuvieron hasta la semana 24. El engrosamiento retinal en la TCO en la semana 24 en comparación con el valor basal fue una variable de eficacia secundaria tanto en el estudio COPERNICUS como en el GALILEO. En ambos estudios, el cambio medio en el engrosamiento retinal desde el valor basal hasta la semana 24 fue estadísticamente significativo favoreciendo a Eylia.
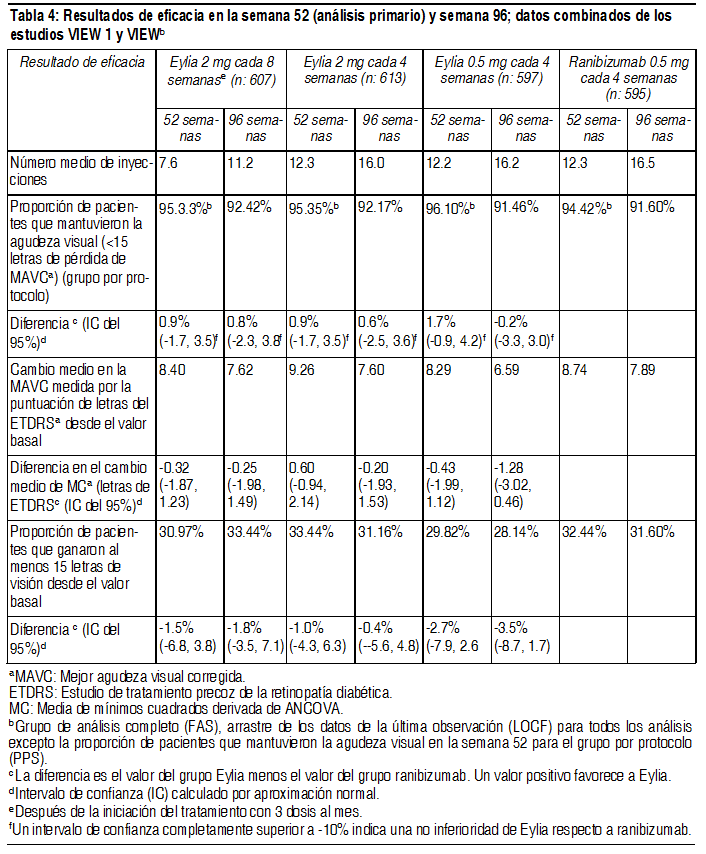
Ver Tabla
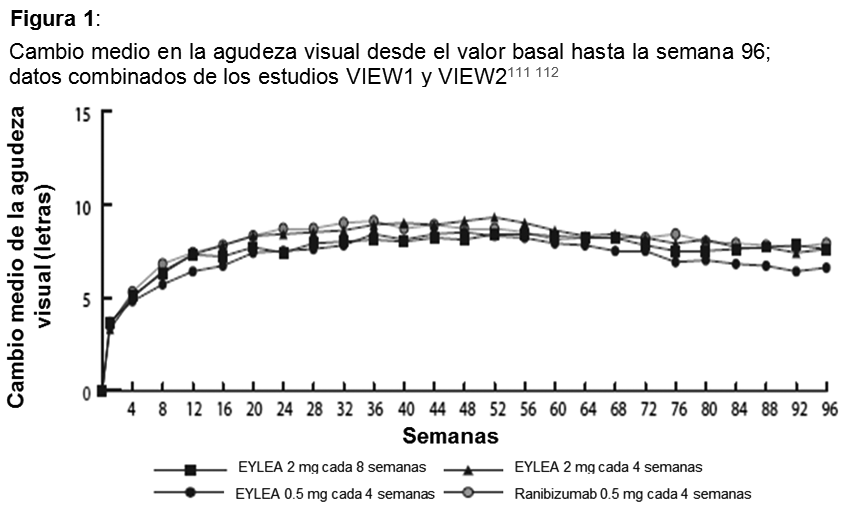
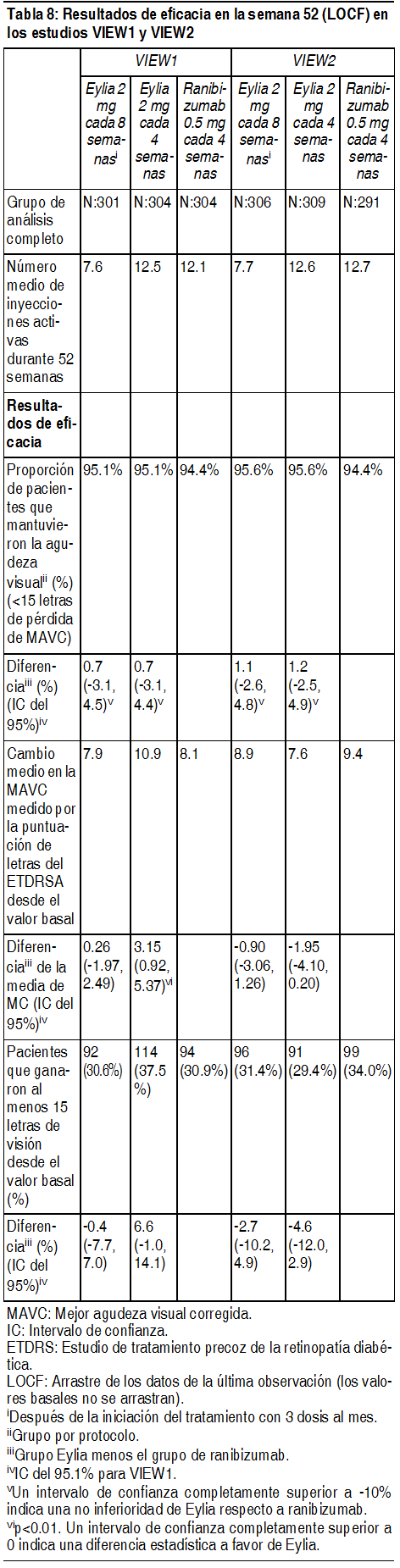
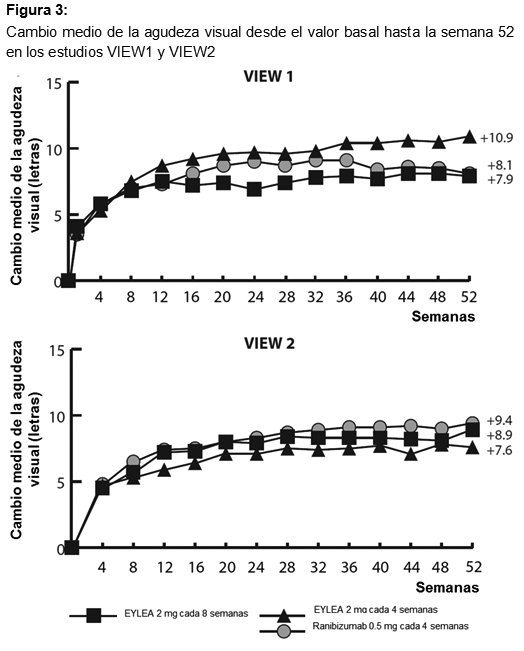
Eficacia clínica: Degeneración macular asociada a la edad (AMD húmeda) neovascular (húmeda): La eficacia y la seguridad de Eylia se evaluaron en 2 estudios aleatorizados, multicéntricos, con doble enmascaramiento, controlados con principio activo en pacientes con AMD húmeda. Un total de 2412 pacientes fueron tratados y evaluables para eficacia (1817 con Eylia) en los 2 estudios (VIEW1 y VIEW2). En cada estudio, los pacientes fueron asignados aleatoriamente en una relación 1:1:1:1 a 1 de 4 pautas posológicas: 1) Eylia administrado a 2 mg cada 8 semanas después de 3 dosis iniciales mensuales (Eylia 2 mg cada 8 semanas). 2) Eylia administrado a 2 mg cada 4 semanas (Eylia 2 mg cada 4 semanas), 3) Eylia administrado a 0.5 mg cada 4 semanas (Eylia 0.5 mg cada 4 semanas) y 4) Ranibizumab administrado a 0.5 mg cada 4 semanas (ranibizumab 0.5 mg cada 4 semanas). Los pacientes tenían edades entre 49 y 99 años, con una media de 76 años. En el segundo año de los estudios, los pacientes siguieron recibiendo las concentraciones de dosis a las que fueron inicialmente asignados en forma aleatoria, pero con un cronograma de dosis modificado guiado por una evaluación de resultados visuales y anatómicos con un intervalo de dosis máximo de 12 semanas definido por el protocolo. Durante el segundo año de los estudios, el 90% de los pacientes originalmente tratados con Eylia 2 mg cada 8 semanas recibió 6 dosis o menos y el 72% recibió 4 dosis o menos entre aquellos pacientes que completaron el segundo año de los estudios. En ambos estudios, la variable primaria de eficacia fue la proporción de pacientes en el grupo por protocolo que mantenían visión, definida como la pérdida de menos de 15 letras de agudeza visual en la semana 52, en comparación con el valor basal. En el estudio VIEW1, en la semana 52, el 95.1% de pacientes del grupo de tratamiento Eylia 2 mg cada 8 semanas, el 95.1% de pacientes en el grupo de tratamiento Eylia 2 mg cada 4 semanas y el 95.9% de pacientes en el grupo de tratamiento Eylia 0.5 mg cada 4 semanas mantuvieron la visión, en comparación con el 94.4% de pacientes en el grupo ranibizumab 0.5Q4. En todos los grupos de tratamiento con Eylia se demostró equivalencia clínica y no inferioridad con el grupo ranibizumab 0.5 mg cada 4 semanas. En el estudio VIEW2, en la semana 52, el 95.6% de pacientes del grupo de tratamiento Eylia 2 mg cada 8 semanas, el 95.6% de pacientes en el grupo de tratamiento Eylia 2 mg cada 4 semanas y el 96.3% de pacientes en el grupo de tratamiento Eylia 0.5 mg cada 4 semanas mantuvieron la visión, en comparación con el 94.4% de pacientes en el grupo ranibizumab 0.5 mg cada 4 semanas. En todos los grupos de tratamiento con Eylia se demostró equivalencia clínica y no inferioridad con el grupo ranibizumab 0.5 mg cada 4 semanas. Los resultados detallados del análisis combinado de ambos estudios se presentan en la tabla y la figura siguientes.
Ver Tabla
Ver Tabla
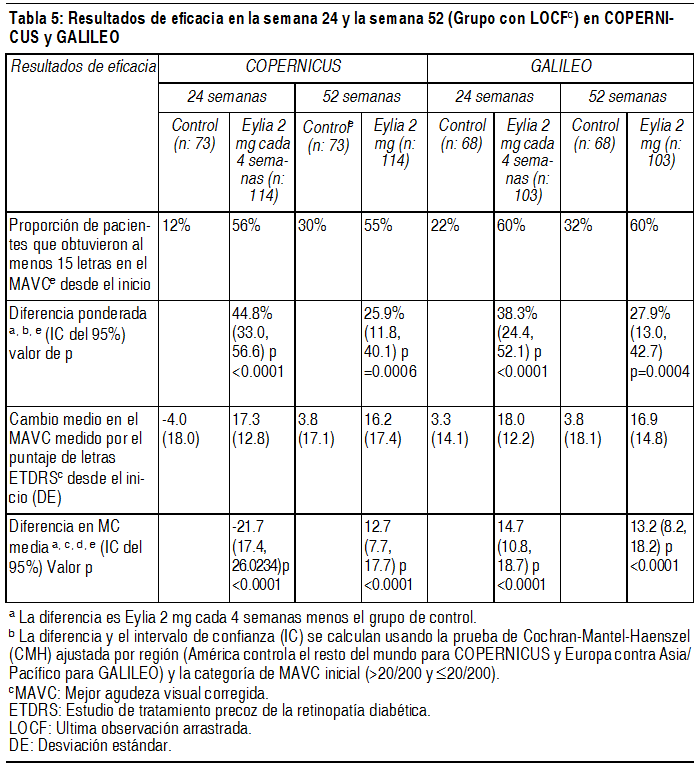
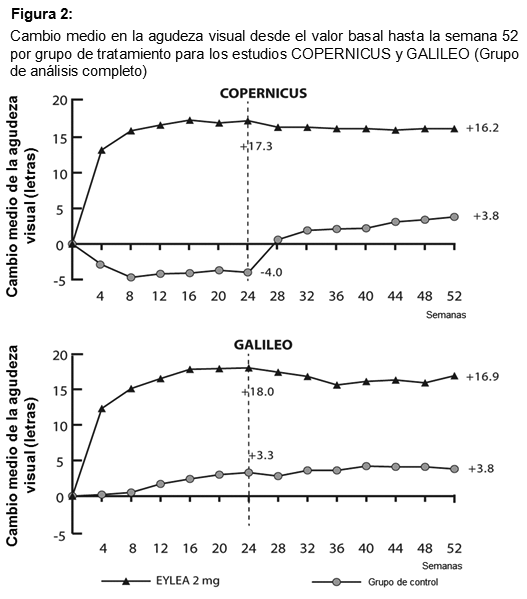
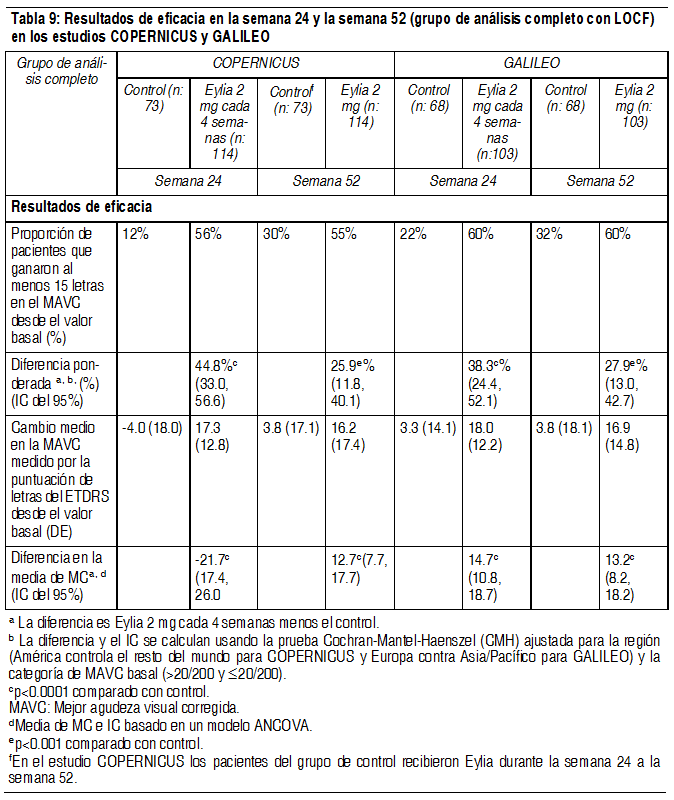
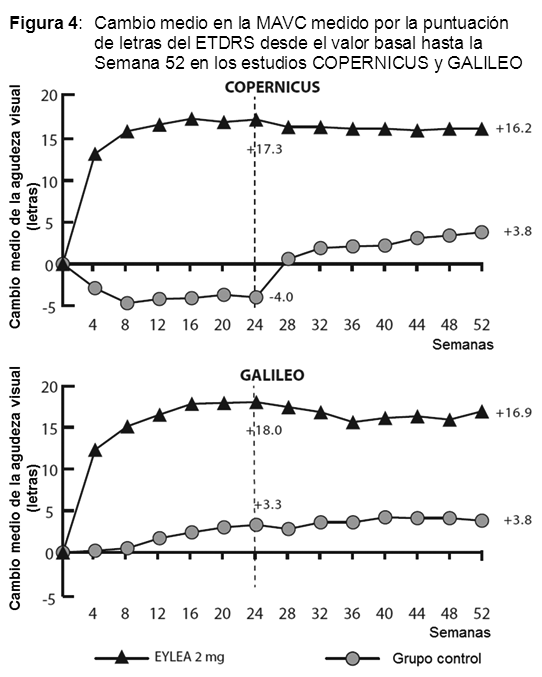
Fueron evidentes disminuciones del área media de la NVC en todos los grupos de dosis en ambos estudios. En el análisis de datos combinados de los estudios VIEW1 y VIEW2, todas las dosis (2 mg cada 8 semanas, 2 mg cada 4 semanas y 0.5 mg cada 4 semanas) de Eylia presentaron cambios clínicamente significativos desde el valor basal de la variable secundaria preespecificada de eficacia del Cuestionario de Funcionamiento Visual del Instituto Nacional del Ojo (NEI VFQ-25). La magnitud de estos cambios fue similar a la observada en los estudios publicados, que corresponde a una ganancia de 15 letras en la mejor agudeza visual corregida (MAVC). No se encontraron diferencias clínicamente significativas entre Eylia y el producto de referencia ranibizumab en los cambios de las subescalas y puntuaciones totales del NEI VFQ-25 (actividades de cerca, actividades de lejos y la dependencia específica de la visión) en la semana 52 con respecto al basal. En el segundo año de los estudios, la eficacia en general se mantuvo hasta la última evaluación en la semana 96. En el período de 2 años, los pacientes en el grupo de Eylia 2 mg cada 8 semanas recibieron un promedio de 11.2 dosis y los pacientes en el grupo de ranibizumab recibieron un promedio de 16.5 dosis. Los resultados de eficacia en todos los subgrupos evaluables (por ejemplo, edad, sexo, raza, agudeza visual inicial, tipo de lesión, tamaño de la lesión) en cada estudio y en el análisis combinado fueron congruentes con los resultados en las poblaciones generales. Pacientes geriátricos: En los estudios clínicos de AMD húmeda, aproximadamente el 89% (1616/1817) de los pacientes aleatorizados a tratamiento con Eylia tenía 65 años o más y aproximadamente el 63% (1139/1817) tenía 75 años o más. Edema macular posterior a la oclusión de la vena central de la retina (OVCR): La seguridad y la eficacia de Eylia se evaluaron en dos estudios aleatorizados, multicéntricos, a doble ciego, controlados con simulación en pacientes con edema macular posterior a OVCR. Un total de 358 pacientes fueron tratados y evaluables para la eficacia (217 con EYLIA) en los dos estudios COPERNICUS y GALILEO. En ambos estudios, los pacientes fueron aleatorizados en una proporción de 3:2 a ya sea 2 mg de Eylia administrados cada 4 semanas (2 mg cada 4 semanas) o al grupo de control que recibían inyecciones simuladas cada 4 semanas por un total de 6 inyecciones. Después de 6 inyecciones mensuales, los pacientes recibieron tratamiento sólo si cumplían con los criterios de retratamiento especificados previamente, excepto los pacientes en el grupo de control en el estudio GALILEO que continuaron recibiendo la simulación (control para el control). Las edades de los pacientes oscilaban entre los 22 y 89 años con un promedio de 64 años. En ambos estudios, el criterio de valoración primaria de eficacia fue la proporción de pacientes que ganaron al menos 15 letras en la MAVC en la semana 24 en comparación con el valor basal. El cambio en la agudeza visual en la semana 24 en comparación con el inicio fue una variable de eficacia secundaria en ambos estudios COPERNICUS y GALILEO. La diferencia entre los grupos de tratamiento fue estadísticamente significativa a favor de Eylia en ambos estudios. Se mantuvo la diferencia estadísticamente significativa hasta la semana 52. En la Tabla y la Figura siguientes se muestran los resultados detallados de los análisis de ambos estudios.
Ver Tabla
Ver Tabla
Los efectos del tratamiento en todos los subgrupos evaluables (por ejemplo, edad, sexo, raza, agudeza visual basal, estado de perfusión retinal, duración de OVCR) en cada estudio fueron en general congruentes con los resultados en las poblaciones generales. Pacientes geriátricos: En los estudios OVCR, aproximadamente el 52% (112/217) de los pacientes aleatorizados al tratamiento con Eylia tenía 65 años de edad o más, y aproximadamente el 18% (38/217) tenía 75 años de edad o más. Propiedades farmacocinéticas: Eylia se administra directamente en el vítreo para ejercer efectos locales en el ojo. Absorción/distribución: Aflibercept es absorbido lentamente desde el ojo en la circulación sistémica después de la administración intravítrea y se observa predominantemente en la circulación sistémica como un complejo estable, inactivo con VEGF; sin embargo, sólo "aflibercept libre" puede unirse al VEGF endógeno. En un subestudio farmacocinético con muestreo frecuente en pacientes con AMD, las concentraciones plasmáticas máximas de aflibercept libre (Cmáx sistémica) fueron bajas, con una media de aproximadamente 0.02 microgramos/ml (rango 0 a 0.054) en 1 a 3 días después de una inyección intravítrea de 2 mg, y no eran detectables 2 semanas después de la administración en casi todos los pacientes. Aflibercept no se acumula en el plasma cuando se administra de forma intravítrea cada 4 semanas. La concentración plasmática máxima media de aflibercept libre es aproximadamente 50 a 500 veces inferior a la concentración de aflibercept requerida para inhibir la actividad biológica del VEGF sistémico un 50% en modelos animales en donde se observaron cambios en la presión arterial luego de que niveles circulares de aflibercept libre alcanzaron aproximadamente 10 microgramos/ml y regresaron al valor basal cuando los niveles cayeron aproximadamente 1 microgramo/ml. Se estima que después de la administración intravítrea de 2 mg a pacientes, la concentración media plasmática máxima de aflibercept libre es más de 100 veces menor que la concentración de aflibercept requerida para la unión semimáxima a VEGF sistémico (2.91 microgramos/ml) en un estudio de voluntarios sanos. Por tanto, son improbables los efectos farmacodinámicos sistémicos como cambios en la presión arterial. Estos resultados farmacocinéticos se confirmaron en un subestudio farmacocinético en pacientes con OVCR (Cmáx media de aflibercept libre en plasma 0.046 microgramos/ml (rango: 0 a 0.081); concentraciones no detectables alcanzadas en 1 semana). Eliminación: No se han realizados estudios de metabolismo ya que Eylia es producto terapéutico basado en proteínas. El aflibercept libre se une a VEGF para formar un complejo estable e inerte. Como con otras proteínas grandes, es de esperar que tanto el aflibercept libre como el no libre se eliminen por catabolismo proteolítico. Pacientes con insuficiencia renal: No se han realizado estudios específicos con Eylia en pacientes con insuficiencia renal. El análisis farmacocinético de pacientes con AMD húmeda en el estudio VIEW2, de los que el 40% tenía insuficiencia renal (24% leve, 15% moderada y 1% severa), no reveló diferencias con respecto a las concentraciones plasmáticas de principio activo después de la administración intravítrea cada 4 u 8 semanas. Se observaron resultados similares en pacientes con OVCR en el estudio GALILEO. Datos preclínicos sobre seguridad: Efectos en los estudios preclínicos sobre la toxicidad a dosis repetidas sólo se observaron a exposiciones sistémicas consideradas muy superiores a la exposición máxima humana después de la administración intravítrea de la dosis clínica propuesta, lo que indica la poca relevancia en el uso clínico. Se observaron erosiones y ulceraciones del epitelio respiratorio en los cornetes nasales de monos tratados con aflibercept por vía intravítrea a exposiciones sistémicas superiores a la exposición máxima humana. La exposición sistémica basada en la Cmáx y el AUC de aflibercept libre eran aproximadamente 200 y 700 veces mayores, respectivamente, cuando se compararon con los valores correspondientes observados en humanos después de una dosis intravítrea de 2 mg. Al nivel sin efecto adverso observable (NOAEL) de 0.5 mg/ojo en monos, la exposición sistémica fue 42 y 56 veces mayor basada en la Cmáx y el AUC, respectivamente. No se han realizado estudios sobre el potencial mutagénico o carcinogénico de aflibercept. Se demostró un efecto de aflibercept en el desarrollo intrauterino en estudios de desarrollo embriofetal en conejas gestantes con administración intravenosa (3 a 60 mg/kg) así como con administración subcutánea (0.1 to 1 mg/kg). El NOAEL materno fue a la dosis de 3 mg/kg o 1 mg/kg, respectivamente. No se identificó un NOAEL de desarrollo. A la dosis de 0.1 mg/kg, las exposiciones sistémicas basadas en la Cmáx y el AUC acumulada de aflibercept libre eran aproximadamente 17 y 10 veces mayores, respectivamente, cuando se compararon con los valores correspondientes observados en humanos después de una dosis intravítrea de 2 mg. Los efectos sobre la fertilidad femenina y masculina se evaluaron, como parte de un estudio de 6 meses en monos, con administración intravenosa de aflibercept a dosis en el rango de 3 a 30 mg/kg. Se observaron menstruaciones ausentes o irregulares asociadas a alteraciones en los niveles de hormonas femeninas de la reproducción y cambios en la motilidad y morfología de los espermatozoides a todos los niveles de dosis. En base a la Cmáx y al AUC de aflibercept libre observados con la dosis intravenosa de 3 mg/kg, las exposiciones sistémicas fueron aproximadamente 4900 y 1500 veces mayores, respectivamente, que la exposición observada en humanos después de una dosis intravítrea de 2 mg. Todos los cambios fueron reversibles.
|
|
Posología:
Eylia es para inyección intravítrea. Sólo debe ser administrado por un médico cualificado con experiencia en la administración de inyecciones intravítreas. Pauta posológica: Degeneración macular asociada a la edad (AMD húmeda) neovascular (húmeda): La dosis recomendada de Eylia es de 2 mg de aflibercept (equivalente a 50 microlitros). El tratamiento con Eylia se inicia con una inyección mensual para 3 dosis consecutivas, seguida por una inyección cada 2 meses. Sin embargo, luego de los primeros 12 meses de tratamiento con EYLIA, el intervalo del tratamiento se podría ampliar hasta cada 3 meses (12 semanas) en base a los resultados visuales y anatómicos. En este caso, el programa de monitorización se determinará según criterio médico y puede ser más frecuente que el programa de administración de las inyecciones. Aunque Eylia puede administrarse con una frecuencia de 1 vez al mes, no se ha demostrado una eficacia adicional cuando Eylia es administrado cada 4 semanas con respecto a la administración cada 8 semanas. Edema macular posterior a la oclusión de la vena central de la retina (OVCR): La dosis recomendada de Eylia es de 2 mg de aflibercept (equivalente a 50 microlitros), administrada a través de inyección intravítrea 1 vez al mes. Información adicional sobre poblaciones especiales: Pacientes con insuficiencia hepática y/o renal: No se han realizado estudios específicos en pacientes con insuficiencia hepática y/o renal con Eylia. Los datos disponibles no indican la necesidad de ajustar la dosis con Eylia en estos pacientes (ver sección Propiedades farmacodinámicas). Pacientes de edad avanzada: No es necesaria ninguna consideración especial. Niños y adolescentes: La AMD húmeda no se presenta en niños y adolescentes. Por tanto, la seguridad y la eficacia de Eylia no se ha estudiado en estos grupos de edad. Forma de administración: Las inyecciones intravítreas deben realizarse conforme a los estándares médicos y las directrices aplicables por un médico cualificado con experiencia en la administración de inyecciones intravítreas. En general, tienen que garantizarse anestesia adecuada y asepsia, incluyendo microbicidas tópicos de amplio espectro (por ej., povidona iodada). Se recomienda desinfección quirúrgica de las manos, guantes estériles, campos estériles y un blefaróstato estéril (o equivalente). Inmediatamente después de la inyección intravítrea, se debe monitorizar una posible elevación de la presión intraocular en los pacientes. La monitorización adecuada puede consistir en un control de la perfusión de la cabeza del nervio óptico o tonometría. En caso necesario, debe haber disponible un equipo estéril para paracentesis. Después de la inyección intravítrea se debe instruir a los pacientes para que informen, inmediatamente, sobre cualquier síntoma sugestivo de endoftalmitis (por ej., dolor ocular, enrojecimiento ocular, fotofobia, visión borrosa). Cada jeringa precargada o vial debe utilizarse únicamente para el tratamiento de un único ojo. Después de la inyección, cualquier producto no utilizado debe desecharse.
|
|
Modo de Empleo:
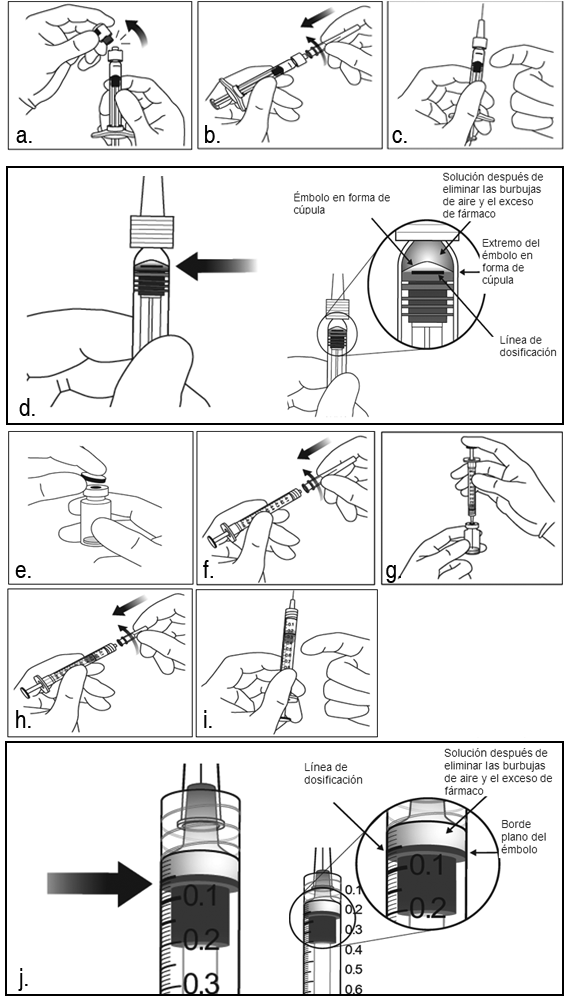
Instrucciones de uso/manipulación: La jeringa precargada y el vial son de un solo uso. Antes de la administración, inspeccione visualmente la solución inyectable. No use la jeringa precargada o el vial si son visibles partículas, turbidez o descoloración. Antes de utilizar, el vial o el envase blíster no abiertos de Eylia pueden conservarse a temperatura ambiente (2ºC/77ºF) durante 24 horas. Después de abrir el vial o el envase blíster, continuar bajo condiciones asépticas. Para la inyección intravítrea se debe usar una aguja hipodérmica de 30 G x 0.5 pulgadas. Jeringa precargada: 1. Cuando esté listo para administrar Eylia, abrir el estuche y sacar el envase blíster esterilizado. Despegue con cuidado el envase blíster, asegurando la esterilidad de su contenido. Mantenga la jeringa en la bandeja estéril hasta que usted esté listo para el ensamblaje. 2. Usando técnica aséptica, saque la jeringa del envase blíster esterilizado. 3. Para quitar el capuchón de la jeringa, sujete la jeringa con una mano mientras que usa la otra mano para agarrar el capuchón de la jeringa con el dedo pulgar e índice. Observación: Quitar (no girar o doblar) el capuchón de la jeringa. 4. Para evitar comprometer la esterilidad del producto, no tire del émbolo. 5. Usando técnica aséptica, enrosque firmemente la aguja hipodérmica en la punta de la jeringa Luer-Lock. 6. Quitar la protección de plástico de la aguja. 7. Sujete la jeringa con la aguja hacia arriba, compruebe que no hay burbujas en la jeringa. Si hay burbujas, golpee suavemente la jeringa con los dedos hasta que las burbujas se desplacen a la parte superior. 8. Para eliminar todas las burbujas y para expulsar el exceso de fármaco, empuje despacio el émbolo hasta alinear la base cilíndrica del émbolo en forma de cúpula con la línea negra de dosificación en la jeringa (equivalente a 50 microlitros). Viales: 1. Quitar la tapa de plástico y desinfectar la parte exterior del tapón de goma del vial. 2. Acoplar la aguja con filtro de 18 G y 5 micras suministrada en el estuche a una jeringa Luer-lock estéril de 1 ml. 3. Introducir la aguja con filtro en el centro del tapón del vial hasta que la aguja esté completamente insertada en el vial y la punta toque el extremo inferior o fondo del vial. 4. Usando técnica aséptica, extraiga todo el contenido del vial de Eylia en la jeringa, manteniendo el vial en una posición vertical, ligeramente inclinado para facilitar la extracción completa. Para impedir la introducción de aire, asegúrese que el bisel de la aguja con filtro está sumergido en el líquido. Continúe inclinando el vial durante la extracción manteniendo el bisel de la aguja con filtro sumergida en el líquido. 5. Asegurarse que el vástago del émbolo se ha retirado suficientemente cuando vacíe el vial para vaciar completamente la aguja con filtro. 6. Quitar la aguja con filtro y desecharla adecuadamente. Nota: La aguja con filtro no debe utilizarse para la inyección intravítrea. 7. Usando técnica aséptica, enrosque firmemente una aguja hipodérmica de 30 G x 0.5 pulgadas a la punta de la jeringa Luer-lock. 8. Cuando esté listo para administrar Eylia, quitar la protección de plástico de la aguja. 9. Sujete la jeringa con la aguja hacia arriba, compruebe que no hay burbujas en la jeringa. Si hay burbujas, golpee suavemente la jeringa con los dedos hasta que las burbujas se desplacen a la parte superior. 10. Eliminar todas las burbujas y expulsar el exceso de fármaco empujando despacio el émbolo, de modo que su extremo se alinee con la línea que marca 0.05 ml en la jeringa.
Ver Tabla
|
|
Efectos Colaterales:
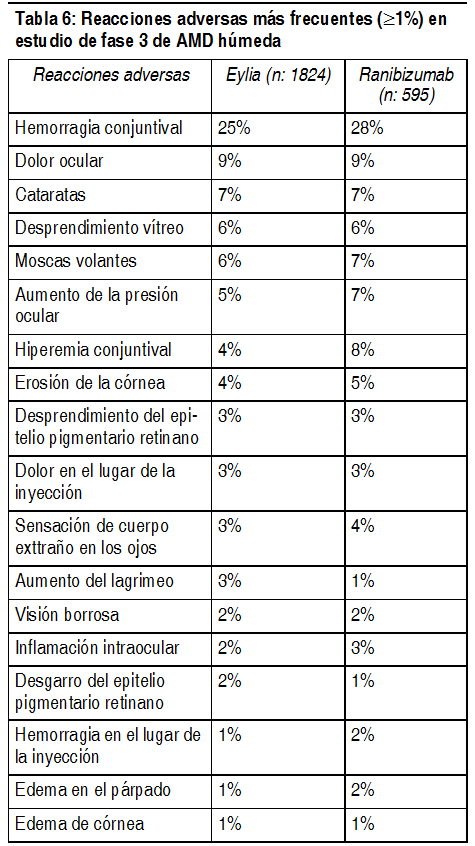
Resumen del perfil de seguridad: Un total de 2099 pacientes tratados con Eylia constituyeron la población de seguridad en los cuatro estudios de fase III. Entre estos, 1498 pacientes fueron tratados con la dosis recomendada de 2 mg. Población AMD húmeda: Un total de 1824 pacientes constituyeron la población de seguridad en los 2 estudios clínicos de fase III con hasta 96 semanas de exposición a EYLIA, de los cuales 1223 pacientes fueron tratados con la dosis de 2 mg. Se han presentado eventos adversos serios relacionados con el procedimiento de inyección en menos de 1 inyección intravítrea de cada 1000 con Eylia e incluyeron endoftalmitis, catarata traumática y aumento transitorio de la presión intraocular (ver sección Advertencias y precauciones especiales de empleo). Los eventos adversos más frecuentes (en al menos el 5% de los pacientes tratados con Eylia) fueron hemorragia conjuntival (26.7%), dolor ocular (10.3%), desprendimiento vítreo (8.4%), cataratas (7.9%), moscas volantes (7.6%) y aumento de la presión intraocular (7.2%). Estas reacciones adversas se presentaron con una incidencia similar en el grupo de tratamiento con ranibizumab. Población OVCR: Un total de 275 pacientes tratados con al menos una dosis de Eylia constituyeron la población de seguridad en los 2 estudios de fase III con una exposición de hasta 52 semanas. Las reacciones adversas graves relacionadas con el procedimiento de la inyección ocurrieron en 2 de 2052 inyecciones intravítreas con Eylia e incluyeron endoftalmitis (ver sección Advertencias y precauciones especiales de empleo) y desprendimiento vítreo. Las reacciones adversas más frecuentes (en al menos 5% de los pacientes tratados con Eylia) fueron hemorragia conjuntival (14.2%), aumento de la presión intraocular (13.5%), dolor ocular (13.1%), desprendimiento vítreo (6.5%) y moscas volantes (5.5%). Lista tabulada de reacciones adversas: Datos de seguridad integrados de la población AMD húmeda y OVCR: Los datos de seguridad descritos a continuación incluyen todas las reacciones adversas (serias y no serias) de los estudios de fase III de AMD húmeda y/o OVCR con una posibilidad razonable de causalidad con el procedimiento de inyección o con el medicamento. Las reacciones adversas se listan con un sistema de clasificación de órgano o sistema y frecuencia usando el siguiente criterio: Muy frecuentes ( ³ 1/10 pacientes); Frecuentes ( ³ 1/100 a <1/10 pacientes); Poco frecuentes ( ³ 1/1000 a <1/100 pacientes), Muy poco frecuentes ( ³ 1/10000 a < 1/1000 pacientes). Trastornos del sistema inmunológico: Poco frecuentes: Hipersensibilidad. Trastornos oculares: Muy frecuentes: Hemorragia conjuntival. Dolor ocular. Frecuentes: Desgarro del epitelio pigmentario retiniano*. Desprendimiento del epitelio pigmentario retiniano*. Cataratas. Cataratas nucleares. Cataratas subcapsulares. Erosión corneal. Abrasión corneal. Aumento de la presión intraocular. Visión borrosa. Moscas volantes. Edema corneal. Desprendimiento vítreo. Dolor en el lugar de la inyección. Sensación de cuerpo extraño en los ojos. Aumento del lagrimeo. Edema palpebral. Hemorragia en el lugar de la inyección. Hiperemia conjuntival. Hiperemia ocular. Poco frecuentes: Endoftalmitis**. Desprendimiento retiniano. Desgarro retiniano. Iridociclitis. Cataratas corticales. Opacidades ienticulares. Defecto del epitelio corneal. Turbidez del humor acuoso de la cámara anterior. Muy poco frecuentes: Vitritis. Uveítis. Iritis. Hipopión. *Condiciones conocidas asociadas con AMD húmeda. Observadas sólo en los estudios de AMD húmeda. **Endoftalmitis de cultivo positivo y cultivo negativo. Además, 157 pacientes de AMD húmeda fueron tratados durante 44 meses en una extensión a largo plazo de los estudios de fase I y fase II. El perfil de seguridad fue consistente con el observado en los estudios de fase III de AMD húmeda. Descripción de las reacciones adversas seleccionadas: Eventos tromboembólicos arteriales: Eventos tromboembólicos arteriales (ETA) son eventos adversos potencialmente relacionados con la inhibición sistémica del VEGF. Hay un riesgo teórico de ETA después del uso intravítreo de inhibidores del VEGF. Los ETA, definidos por criterios de Colaboración de Ensayistas Antiplaquetarios (APTC), incluyen infarto de miocardio no fatal, accidente cerebrovascular no fatal o muerte vascular (incluyendo muertes de causa desconocida). La incidencia en los estudios de AMD húmeda VIEW1 y VIEW2 durante las 96 semanas de duración del estudio fue 3.3% (60 de 1824) en el grupo combinado de pacientes tratados con Eylia, en comparación con 3.2% (19 de 595) en los pacientes tratados con ranibizumab (ver sección Propiedades farmacodinámicas/Eficacia clínica). La incidencia de los ETA en los estudios GALILEO y COPERNICUS de OVCR en la duración del estudio de 52 semanas fue 0.4% (1 de 275) pacientes tratados con al menos una dosis de EYLIA en comparación con 2.4% (2 de 85) en el grupo de pacientes que reciben sólo el tratamiento simulado. Inmunogenicidad: Como con todas las proteínas terapéuticas, hay un potencial de inmunogenicidad con Eylia. La inmunogenicidad se evaluó en muestras de suero. Los datos de inmunogenicidad reflejan el porcentaje de pacientes cuyos resultados analíticos se consideraron positivos para anticuerpos contra Eylia en inmunoensayos y son altamente dependientes de la sensibilidad y especificidad de los ensayos. En los estudios de fase III de AMD húmeda y OVCR, la incidencia pretratamiento de inmunorreactividad a Eylia fue menos que aproximadamente 3% en todos los grupos de tratamiento. Después de la administración de Eylia durante 96 semanas (AMD húmeda), o 52 semanas (OVCR) se detectaron anticuerpos contra Eylia en un porcentaje similar de pacientes. No hubo diferencias de eficacia o seguridad entre los pacientes con o sin inmunorreactividad tanto en los estudios de AMD húmeda como en los de OVCR. En general, el riesgo de inmunogenicidad significativa con Eylia parece ser muy bajo.
|
|
Contraindicaciones:
Infección ocular o periocular. Inflamación intraocular activa severa. Hipersensibilidad conocida a aflibercept o a alguno de sus excipientes.
|
|
Advertencias:
Advertencias y precauciones especiales de empleo: Endoftalmitis: Las inyecciones intravítreas, incluyendo las de Eylia, se han asociado a endoftalmitis (ver sección Efectos cloaterales). Siempre que se administre Eylia se debe emplear técnica de inyección aséptica adecuada. Se debe instruir a los pacientes para que informen inmediatamente cualquier síntoma sugestivo de endoftalmitis y deben tratarse adecuadamente. Aumento de la presión intraocular: Se han observado aumentos de la presión intraocular en los 60 minutos siguientes a una inyección intravítrea, incluida Eylia (ver sección Efectos colaterales). Se ha de tener precaución especial en los pacientes con glaucoma mal controlado. Por lo tanto, en todos los casos se deberá realizar un seguimiento y tratar adecuadamente tanto de la presión intraocular como de la perfusión de la cabeza del nervio óptico.
|
|
Precauciones:
Embarazo y lactancia: Embarazo: No se dispone de datos sobre el uso de aflibercept en mujeres embarazadas. Los estudios en animales han mostrado toxicidad reproductiva después de la administración sistémica (ver sección Datos preclínicos sobre seguridad). Eylia no está recomendado durante el embarazo, a menos que el beneficio potencial supere el posible riesgo para el feto. Mujeres en edad fértil: Las mujeres en edad fértil deberán utilizar un método anticonceptivo eficaz durante el tratamiento. Lactancia: Se desconoce si aflibercept es excretado en la leche materna. No puede excluirse un riesgo para el lactante. Eylia no se recomienda durante la lactancia. Debe tomarse la decisión sobre si interrumpir la lactancia o abstenerse del tratamiento con Eylia. Embarazo: Categoría C del embarazo: No se dispone de estudios adecuados y bien controlados en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción después de la administración sistémica. Eylia sólo debe utilizarse durante el embarazo si el posible efecto beneficioso justifica el riesgo potencial para el feto. Aflibercept produjo toxicidad embriofetal en un estudio de desarrollo embriofetal en conejas gestantes con administración intravenosa (3 a 60 mg/kg). El nivel sin efecto adverso observable (NOAEL) materno fue a la dosis de 3 mg/kg. A esta dosis, las exposiciones sistémicas basadas en la Cmax y el AUC de aflibercept libre eran aproximadamente 2900 y 600 veces mayores, respectivamente, cuando se compararon con los valores correspondientes observados en humanos después de una dosis intravítrea de 2 mg. Mujeres en edad fértil: Las mujeres en edad fértil deberán utilizar un método anticonceptivo eficaz durante el tratamiento. Madres lactantes: Se desconoce si aflibercept es excretado en la leche materna. No puede excluirse un riesgo para el lactante ya que muchos fármacos se excretan en la leche humana. Eylia no se recomienda durante la lactancia. Por tanto, debe decidirse si interrumpir la lactancia o interrumpir el tratamiento con Eylia, teniendo en cuenta la importancia del medicamento para la madre. Efectos sobre la capacidad de conducir o utilizar máquinas: Los pacientes pueden experimentar trastornos visuales temporales después de una inyección intravítrea con Eylia y los exámenes oculares asociados. No deben conducir ni utilizar máquinas hasta que se haya recuperado suficientemente la función visual.
|
|
Interacciones Medicamentosas:
No se han realizado estudios formales de interacción de medicamentos con Eylia.
|
|
Sobredosificación:
En general, fueron bien toleradas dosis de hasta 4 mg a intervalos mensuales en los ensayos clínicos y de 8 mg en casos aislados de sobredosis. La sobredosis con aumento del volumen de inyección puede incrementar la presión intraocular. Por tanto, en caso de sobredosis se debe monitorizar la presión intraocular y, si se considera necesario por el médico responsable, debe iniciarse tratamiento adecuado.
|
|
Incompatibilidades:
Eylia no debe mezclarse con otros medicamentos.
|
|
Conservación:
Conservar en frigorífico (de 2ºC a 8ºC / 36ºF a 46ºF). No congelar. Mantenga la jeringa precargada en su envase blíster y dentro del empaque de cartón externo para protegerla de la luz. Conservar el vial en el embalaje exterior para protegerlo de la luz.
|
|
Observaciones:
Apéndice 1: Presentación de la información sobre reacciones adversas específica para los Estados Unidos: Experiencia en estudios clínicos: Un total de 2042 pacientes tratados con Eylia constituyeron la población de seguridad en los 4 estudios de fase 3. Entre estos, 1441 pacientes se trataron con la dosis recomendada de 2 mg. Degeneración macular relacionada con la edad neovascular (húmeda): Los datos que se describen a continuación reflejan la exposición a Eylia en 1824 pacientes con AMD húmeda, incluidos 1223 pacientes tratados con la dosis de 2 mg, en 2 estudios con doble enmascaramiento, con control activo (VIEW1 y VIEW2) durante 12 meses [ver Estudios clínicos]. Los datos de seguridad que se describen a continuación incluyen todas las reacciones adversas con una posibilidad razonable de causalidad relacionada con el procedimiento de inyección, Eylia, o ranibizumab.
Ver Tabla
Las reacciones adversas graves menos frecuentes informadas en <1% de los pacientes tratados con Eylia fueron desprendimiento de la retina, desgarro de la retina y endoftalmitis. También se ha informado hipersensibilidad en menos del 1% de los pacientes tratados con Eylia. Edema macular posterior a la oclusión de la vena central de la retina: Apéndice 2: Presentación de la información de estudios clínicos específica para los Estados Unidos: Eficacia clínica: Degeneración macular asociada a la edad neovascular (húmeda).
Ver Tabla
Ver Tabla
Fueron evidentes disminuciones del área media de la NVC en todos los grupos de dosis en ambos estudios.
Los resultados de la eficacia en todos los subgrupos evaluables (por ej., edad, sexo, raza, agudeza visual basal, tipo de lesión y tamaño de la lesión) en cada estudio y en el análisis combinado fueron consistentes con los resultados en las poblaciones globales. Edema macular posterior a la Oclusión de la Vena Central de la Retina (OVCR):
Ver Tabla
Ver Tabla
Los efectos del tratamiento en todos los subgrupos evaluables (por ejemplo, edad, sexo, raza, agudeza visual basal, estado de perfusión retinal, duración de OVCR) en cada estudio y en el análisis combinado fueron en general congruentes con los resultados en las poblaciones generales.
|
|
Presentaciones:
Naturaleza y contenido del envase: Jeringas precargadas: Cada estuche incluye 1 envase blíster sellado con 1 jeringa precargada estéril de vidrio de tipo I que contiene un volumen de llenado de 165 microlitros de solución para inyección intravítrea, sellada con un émbolo y un protector elastoméricos de la punta de la jeringa que forma parte de un sistema de cierre con adaptador Luer lock. La jeringa tiene 1 émbolo prefijado y 1 placa de sujeción. Viales: Cada estuche incluye 1 vial de vidrio de tipo I que contiene un volumen de llenado de 278 microlitros de solución para inyección intravítrea con un tapón elastómero de goma y 1 aguja con filtro de 18 G.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}