 |
Composición:
Cada cápsula contiene: 200 mg de Boceprevir. Ingredientes inactivos: Lauril, Sulfato Sódico, Celulosa Microcristalina, Lactosa Monohidrato, Croscarmelosa Sódica, Almidón Progelatinizado, Gelatina, Dióxido de Titanio, Oxido de Hierro amarillo, Oxido de Hierro Rojo y Estearato de Magnesio.
|
|
Descripción:
Victrelis (boceprevir) es un inhibidor de la proteasa serina de la proteína no estructural 3 (NS3) del virus de la hepatitis (HCV). Boceprevir tiene el siguiente nombre químico: (1R,5S)-N-[3-amino-1-(ciclobutilmetil)-2,3-dioxo-propil]-3-[2(S[[[(1.1-dimetiletil)amino]carbonil]amino]-3.3-dimetil-1-oxobutil]-6.6-dimetil-3-azabiciclo[3.1.0]hexan-2(S)-carboxamida. La fórmula molecular es C27H45N5O5 y su peso molecular es 519.7. Boceprevir tiene la siguiente fórmula estructural:
Ver Tabla
Boceprevir se fabrica como una mezcla aproximadamente equitativa de 2 diastereómeros. Boceprevir es un polvo amorfo de color blanco a blanquecino. Es libremente soluble en metanol, etanol e isopropanol, y ligeramente soluble en agua. Las cápsulas de 200 mg de Victrelis están disponibles en forma de cápsulas de gelatina dura para administración oral. Farmacología clínica: Mecanismo de acción: Victrelis es un fármaco antiviral de acción directa contra el virus de la hepatitis C (véase Microbiología). Farmacodinámica: Evaluación del efecto de Victrelis en el intervalo QTc: El efecto de boceprevir de 800 mg y 1200 mg en el intervalo QTc se evaluó en un estudio completo de QT, randomizado, de dosis múltiples, controlado con placebo y principio activo (moxifloxacina de 400 mg), de diseńo cruzado de 4 vías realizado en 36 sujetos sanos. En el estudio con capacidad demostrada para detectar efectos pequeńos, el límite superior del intervalo de confianza (CI) unilateral del 95% para el intervalo QTc más grande ajustado al placebo y corregido según la basal, de acuerdo con el método de corrección individual (QTcI), fue inferior a 10 ms, el umbral para los asuntos reglamentarios. La dosis de 1200 mg produce un aumento en la exposición máxima de boceprevir de aproximadamente el 15%, que probablemente no cubra las exposiciones debido a la coadministración con fuertes inhibidores del CYP3A4 o al uso en pacientes con insuficiencia hepática severa. Sin embargo, a las dosis estudiadas en el estudio completo de QT, no se identificó ninguna relación aparente entre la concentración y QT. Por ende, no se espera un efecto en QTc en un marco de exposición más elevado. Farmacocinética: Las cápsulas de Victrelis contienen una mezcla 1:1 de 2 diastereómeros, SCH534128 y SCH534129. En el plasma, la proporción de los diastereómeros cambia a 2:1, en favor del diastereómero activo, SCH534128. Las concentraciones plasmáticas de boceprevir descritas a continuación están compuestas por ambos diastereómeros, SCH534128 y SCH534129, salvo que se especifique lo contrario. En participantes sanos que recibieron 800 mg de boceprevir solo, tres veces al día, la exposición al fármaco se caracterizó por un AUC( · ) de 5408 ng x h por mL (n=71), una Cmáx. de 1723 ng x h por mL (n=71) y una Cmín. de 88 ng x h por mL (n=71). Los resultados farmacocinéticos fueron similares entre los participantes sanos y los participantes infectados por HCV. Absorción: Boceprevir se absorbió luego de la administración oral con un Tmáx medio de 2 horas. El AUC, la Cmáx. y la Cmín. en equilibrio dinámico aumentaron de una manera menor que proporcional a la dosis y las exposiciones individuales se superpusieron considerablemente a las dosis de 800 mg y 1200 mg, lo cual sugirió una menor absorción con dosis mayores. La acumulación es mínima (0.8 a 1.5 veces) y el equilibrio dinámico farmacocinético se logra después de aproximadamente 1 día de la administración de dosis de 3 veces al día. No se ha estudiado la biodisponibilidad absoluta de boceprevir. Efectos de los alimentos en la absorción oral: Victrelis se deberá administrar con alimentos. Los alimentos mejoraron la exposición de boceprevir hasta en un 65% a la dosis de 800 mg 3 veces al día, en relación con la administración en ayunas. La biodisponibilidad de boceprevir fue similar independientemente del tipo de comida (por ejemplo, alto contenido graso frente a bajo contenido graso) o si se tomó 5 minutos antes de comer, durante una comida o inmediatamente después de finalizar la comida. Por lo tanto, Victrelis se puede tomar independientemente del tipo de comida o el momento de la comida. Distribución: Boceprevir tiene un volumen de distribución aparente medio (Vd/F) de aproximadamente 772 L en equilibrio dinámico en los participantes sanos. La unión a las proteínas plasmáticas humanas es aproximadamente del 75% luego de 1 dosis única de boceprevir de 800 mg. Boceprevir se administra como una mezcla aproximadamente equivalente de 2 diastereómeros, SCH534128 y SCH534129, que se interconvierten rápidamente en el plasma. El diastereómero predominante, SCH534128, es farmacológicamente activo y el otro diastereómero es inactivo. Metabolismo: Los estudios in vitro indican que boceprevir se metaboliza principalmente a través de la vía mediada por la aldocetorreductasa (AKR) a metabolitos con cetonas reducidas que son inactivos contra el HCV. Después de 1 dosis oral única de 800 mg de 14C-boceprevir, los metabolitos circulantes más abundantes fueron una mezcla diasteriomérica de metabolitos con cetonas reducidas con una exposición media aproximadamente 4 veces mayor que aquella de boceprevir. Boceprevir también experimenta, en menor grado, un metabolismo oxidativo mediado por el CYP3A4/5. Interacciones farmacológicas: Se llevaron a cabo estudios de interacción farmacológica con boceprevir y fármacos que probablemente se coadministren o fármacos comúnmente utilizados como pruebas para las interacciones farmacocinéticas. Los efectos de la coadministración de boceprevir en el AUC, la Cmáx. y la Cmín se resumen en la Tabla 6 (efectos de los fármacos coadministrados sobre boceprevir) y la Tabla 7 (efectos de boceprevir sobre los fármacos coadministrados).
Ver Tabla
Ver Tabla
Eliminación: Boceprevir se elimina con un valor medio de la vida media (t˝) plasmática de aproximadamente 3,4 horas, Boceprevir tiene una depuración corporal total media (CL/F) de aproximadamente 161 L por h. Luego de una dosis oral única de 800 mg de 14C-boceprevir, aproximadamente el 79% y el 9% de la dosis se excretó en las heces y la orina, respectivamente, eliminándose aproximadamente el 8% y el 3% del carbono radioactivo administrado en forma de boceprevir en las heces y la orina. La información indica que boceprevir se elimina principalmente a través del hígado. Poblaciones especiales: Insuficiencia hepática: La farmacocinética de boceprevir se estudió en participantes adultos no infectados por el HCV con función hepática normal, insuficiencia hepática leve (puntuación 5 a 6 en la escala Child-Pugh), moderada (puntuación 7 a 9 en la escala Child-Pugh) y severa (puntuación 10 a 12 en la escala Child-Pugh) luego de una dosis única de 400 mg de Victrelis, El AUC medio del diastereómero activo de boceprevir (SCH534128) fue mayor en un 32% y 45% en los participantes con insuficiencia hepática moderada y severa, respectivamente, en relación con los participantes con función hepática normal, Los valores medios de la Cmáx, para SCH534128 fueron mayores en un 28% y 62% en los participantes con insuficiencia hepática moderada y severa, respectivamente. Los participantes con insuficiencia hepática leve tuvieron una exposición del SCH534128 similar a los participantes con función hepática normal. Se prevé una magnitud similar del efecto para boceprevir. No se recomienda el ajuste de la dosis de Victrelis en los pacientes con insuficiencia hepática [véase Uso en poblaciones específicas]. Véase el Inserto del Empaque de peginterferón alfa para obtener información sobre las contraindicaciones para los pacientes con descompensación hepática. Insuficiencia renal: La farmacocinética de boceprevir se estudió en participantes no infectados por el HCV con nefropatía de fase terminal (ESRD) que requirieron hemodiálisis luego de una dosis única de 800 mg de Victrelius, El AUC medio de boceprevir fue menor en un 10% en los participantes con ESRD que requirieron hemodiálisis en relación con los participantes con función renal normal, La hemodiálisis eliminó menos del 1% de la dosis de boceprevir. No se requiere el ajuste de la dosis de Victrelis en pacientes con cualquier grado de insuficiencia renal. Género: El análisis farmacocinético de la población de Victrelis indicó que el género no tuvo un efecto aparente en la exposición. Raza: El análisis farmacocinético de la población de Victrelis indicó que la raza no tuvo un efecto aparente en la exposición. Edad: El análisis farmacocinético de la población de Victrelis mostró que la exposición de boceprevir no fue diferente entre los participantes de 19 a 65 ańos de edad. Microbiología: Mecanismo de acción: Boceprevir es un inhibidor de la proteasa NS3/4A del HCV que es necesaria para la descomposición proteolítica de la poliproteína codificada del HCV en formas maduras de las proteínas NS4A, NS4B, NS5A y NS5B. Boceprevir se une de manera covalente, aunque reversible, a la serina (S139) del sitio activo de la proteasa NS3 a través de un grupo funcional (alfa)-cetoamida para inhibir la replicación viral en las células huéspedes infectadas por el HCV. En un ensayo bioquímico, boceprevir inhibió la actividad de las enzimas proteasas NS3/4A recombinantes del HCV de genotipo 1a y 1b, con valores de Ki de 14 nM para cada subtipo. Actividad en cultivos celulares: Los valores de la EC50 y la EC90 correspondientes a boceprevir frente a un replicón del HCV creado a partir de una cepa aislada única de genotipo 1b fueron aproximadamente de 200 nM y 400 nM, respectivamente, en un ensayo de cultivo celular de 72 horas. La actividad anti-HCV del cultivo celular con boceprevir fue aproximadamente 2 veces menor en un replicón del HCV proveniente de una cepa aislada única de genotipo 1a, en relación con el replicón proveniente de una cepa aislada de genotipo 1b. En ensayos del replicón, boceprevir tuvo una actividad aproximadamente 2 veces menor frente a una cepa aislada de genotipo 2a, en relación con las cepas aisladas del genotipo 1a y 1b del replicón. En un ensayo bioquímico, boceprevir tuvo una actividad aproximadamente 3 y 2 veces menor frente a las proteasas NS3/4A provenientes de cepas aisladas únicas representativas de los genotipos 2 y 3a del HCV, respectivamente, en relación con una proteasa NS3/4A proveniente del genotipo 1b. La presencia de suero humano al 50% redujo la actividad anti-HCV del cultivo celular de boceprevir en aproximadamente 3 veces. La evaluación de las combinaciones variables de boceprevir e interferón alfa-2b que produjeron una supresión del 90% del ARN del replicón en cultivo celular mostró aditividad del efecto sin evidencia de antagonismo. Resistencia: En cultivos celulares: La resistencia a boceprevir se caracterizó en ensayos bioquímicos y del replicón del genotipo 1b del HCV. La actividad de boceprevir frente a la proteasa NS3/4A del HCV o el replicón del genotipo 1b se redujo (2 a 6 veces) por las siguientes sustituciones de aminoácidos en el dominio de la proteasa NS3: V36A/I/M, Q41R, T54A/S, V55A/I, R155K/M/Q y V158I. Una reducción de más de 10 veces en la actividad anti-HCV de boceprevir fue propiciada por las sustituciones R155G/I/T y A156S/T/V. Las sustituciones V55I y D168N no redujeron la sensibilidad de boceprevir. Las siguientes sustituciones de aminoácidos dobles confirieron más de 10 veces la sensibilidad reducida a boceprevir: V55A + I170V, T54S + R155K, R155K + D168N, D168N y R155T+D168N y V36M + R155K. La actividad de boceprevir contra el replicón VHC de genotipo 1b se redujo (2 a 8 veces) por las siguientes sustituciones de aminoácidos en el dominio de la proteasa NS3: V36A / M, Q41R, F43S, T54A/G/S, V55A / I, R155K, V158I, V170M y M175L. Una reducción mayor que 10 veces en la susceptibilidad boceprevir fue conferido por las sustituciones aminoacidicas A156S/T/V, V170A y V36M + R155K. La sustitución D168V única no redujo la sensibilidad a boceprevir. Sustituciones adicionales en el dominio de la proteasa NS3 que no han sido evaluados en el replicón del VHC, pero se ha demostrado reducir la actividad de boceprevir en contra de la proteasa NS3/4A del VHC en un ensayo bioquímico incluyen F43C y R155G/I/M/Q. Resistencia asociada a las sustituciones aminoacídicas para el genotipo 1a y 1b del VHC se observaron en los ensayos clínicos que se presentan en la Tabla 8. En estudios clínicos: Se llevó a cabo un análisis combinado según el tratamiento de la resistencia genotípica en participantes que recibieron cuatro semanas de PegIntron/Rebetol seguidos por VICTRELIS de 800 mg 3 veces al día en combinación con PegIntron/Rebetol en dos estudios de Fase 3, SPRINT-2 y RESPOND-2, Entre los participantes tratados con Victrelis que no alcanzaron una respuesta virológica sostenida y cuyas muestras se analizaron, el 53% tuvo una o más sustituciones específicas post-basales de aminoácidos del dominio de la proteasa NS3 emergentes del tratamiento detectadas por un ensayo de secuenciación basado en la población (Tabla 8). Se observaron patrones similares de sustituciones emergentes del tratamiento en P06086, un ensayo clínico de fase 3 en pacientes con HCC no tratados previamente con infección por genotipo 1 que compara el uso de la ESA a la reducción de la dosis de ribavirina para el tratamiento inicial de la anemia durante el tratamiento con Victrelis en combinación con PegIntron/Rebetol. Se ha demostrado que casi todas estas sustituciones reducen la actividad anti-HCV de boceprevir en ensayos de cultivos celulares o bioquímicos. Entre los sujetos tratados con Victrelis en SPRINT-2 y RESPOND-2 que no alcanzaron una SVR y cuyas muestras post-basales se analizaron, el 31% de los participantes que respondieron a PegIntron/Rebetol, que se definió como una disminución mayor que o igual a 1 log10 en la carga viral en la semana de tratamiento 4 (final del período de introducción de 4 semanas de PegIntron/Rebetol), tuvo sustituciones detectables emergentes del tratamiento, en comparación con el 68% de los participantes con una disminución menor que 1 log10 en la carga viral en la semana de tratamiento 4. No se observaron patrones claros de sustituciones emergentes del tratamiento con boceprevir en el dominio helicasa de NS3 o las regiones de codificación NS4A del genoma del HCV.
Ver Tabla
Persistencia de las sustituciones asociadas con la resistencia: La información obtenida de un estudio de seguimiento a largo plazo en curso de participantes que no alcanzaron una SVR en pruebas de Fase 2 con Victrelis, con una mediana de duración del seguimiento de aproximadamente 2 ańos, indica que las poblaciones del HCV que albergan ciertas sustituciones post-basales emergentes del tratamiento pueden disminuir con una abundancia relativa en el tiempo. Sin embargo, entre aquellos participantes con información disponible, una o más sustituciones emergentes del tratamiento permanecieron detectables con un ensayo de secuenciación basado en la población en el 25% de los participantes después de 2.5 ańos de seguimiento. Las sustituciones de NS3 más comunes detectadas después de 2.5 ańos de seguimiento fueron T54S y R155K. La falta de detección de una sustitución de acuerdo con un ensayo basado en la población no necesariamente indica que las poblaciones virales que tienen esa sustitución han disminuido a un nivel de referencia que probablemente haya existido antes del tratamiento. Se desconoce el impacto clínico a largo plazo de la emergencia o la persistencia de las sustituciones asociadas con la resistencia a boceprevir. No se dispone de información con respecto a la eficacia de Victrelis entre los participantes que estuvieron expuestos anteriormente a Victrelis o que fracasaron en el tratamiento anterior con un régimen que contenía Victrelis. Efecto de los polimorfismos basales del HCV en la respuesta al tratamiento: Se llevó a cabo un análisis combinado para explorar la asociación entre la detección de polimorfismos basales en aminoácidos de NS3/4A y el resultado del tratamiento en los 2 estudios de Fase 3, SPRINT-2 y RESPOND-2. Se detectaron polimorfismos basales asociados con la resistencia en el 7% de los participantes a través de un método de secuenciación basado en la población, En términos generales, la presencia de estos polimorfismos solos no produjo un impacto en las tasas de SVR en los participantes tratados con Victrelis. Sin embargo, entre los sujetos con una respuesta relativamente deficiente a PegIntron/Rebetol durante el período de introducción de 4 semanas, la eficacia de Victrelis parecía ser menor en aquellos que tenían sustituciones V36M, T54A, T54S, V55A o R155K detectadas en la basal. Los participantes con estos polimorfismos basales y una respuesta menor a PegIntron/Rebetol representaron aproximadamente el 1% del número total de los participantes tratados con Victrelis. Resistencia cruzada: Se ha demostrado que muchas de las sustituciones de aminoácidos de NS3 emergentes del tratamiento detectadas en participantes tratados con Victrelis que no alcanzaron una SVR en las pruebas clínicas de Fase 3 reducen la actividad anti-HCV de otros inhibidores de la proteasa NS3/4A del HCV. No se ha estudiado el impacto de la exposición anterior a Victrelis o el fracaso del tratamiento en la eficacia de otros inhibidores de la proteasa NS3/4A del HCV. No se ha establecido la eficacia de Victrelis en pacientes con antecedentes de exposición a otros inhibidores de la proteasa NS3/4Ş. No se prevé una resistencia cruzada entre Victrelis y los interferones, o Victrelis y ribavirina. Farmacogenómica: Una variante genética cerca del gen que codifica a interferón-lambda-3 (IL28B rs12979860, un cambio de C a T) es un importante factor predictor de la respuesta a PegIntron/Rebetol. La genotipificación de IL28B rs12979860 se efectuó en 653 de 1.048 (62%) participantes en el estudio SPRINT-2 (aquellos que no recibieron tratamiento previo) y en 259 de 394 (66%) participantes en el estudio RESPOND-2 (aquellos que fracasaron en un tratamiento anterior) (véase Estudios clínicos para obtener descripciones de las pruebas).(Entre los participantes que recibieron por lo menos 1 dosis de placebo o Victrelis (población de Intención de tratamiento modificada), las tasas de SVR tendieron a ser menores en los participantes con los genotipos C/T y T/T en comparación con aquellos con el genotipo C/C, particularmente entre los participantes que no recibieron tratamiento previo y recibieron 48 semanas de PegIntron y Rebetol (véase la Tabla 9). Entre aquellos que fracasaron en un tratamiento anterior, los participantes de todos los genotipos parecían tener tasas de SVR mayores con los regímenes que contienen Victrelis. Los resultados de este análisis de subgrupo retrospectivo se deberán observar con precaución debido al pequeńo tamańo de la muestra y las posibles diferencias en las características demográficas y clínicas de la población del subestudio en relación con la población general de la prueba.
Ver Tabla
|
|
Indicaciones:
Victrelis® (boceprevir) está indicado para el tratamiento de la infección crónica por el virus de la hepatitis C de genotipo 1, en combinación con peginterferón alfa y ribavirina, en pacientes adultos (18 ańos de edad y mayores) con enfermedad hepática compensada, incluso cirrosis, que no recibieron tratamiento previo o que han fracasado en una terapia anterior con interferón y ribavirina, incluyendo respuesta anterior nula, respuesta parcial y recaída (véase Estudios clínicos). Los siguientes puntos se deberán considerar cuando se inicie la administración de Victrelis para el tratamiento de la infección crónica por hepatitis C: Victrelis no debe utilizarse como monoterapia y sólo se deberá usar en combinación con peginterferón alfa y ribavirina. La eficacia de Victrelis no se ha estudiado en pacientes que han fracasado en una terapia anterior con un régimen de tratamiento que incluye Victrelis u otros inhibidores de la proteasa NS3/4A del HCV. Los pacientes poco sensibles a interferón que recibieron tratamiento con Victrelis en combinación con peginterferón alfa y ribavirina tienen una menor probabilidad de lograr una respuesta virológica sostenida (SVR) y una mayor tasa de detección de sustituciones asociadas con la resistencia después del fracaso del tratamiento, en comparación con los pacientes con una mayor respuesta a peginterferón alfa y ribavirina (véase Microbiología y Estudios clínicos).
|
|
Propiedades:
Estudios clínicos: La eficacia de Victrelis como un tratamiento para la infección crónica por el virus de la hepatitis C (genotipo 1) se evaluó aproximadamente en 1.500 participantes adultos que no recibieron tratamiento previo (estudio SPRINT-2) o que habían fracasado en una terapia anterior con peginterferón alfa y ribavirina (estudio RESPOND-2) en estudios clínicos de Fase 3. Participantes que no recibieron tratamiento previo: El estudio SPRINT-2 fue un estudio randomizado, a doble ciego, controlado con placebo que comparó dos regímenes terapéuticos de Victrelis de 800 mg por vía oral 3 veces al día en combinación con PR [PegIntron alfa-2b de 1.5 microgramos por kg a la semana por vía subcutánea y dosis basada en el peso con Rebetol (600-1.400 mg al día por vía oral dividida en 2 veces al día)] con PR solo en participantes adultos que padecían de infección crónica por el virus de la hepatitis C (genotipo 1 del HCV) con niveles detectables del ARN del HCV y que no recibieron tratamiento previo con la terapia con interferón alfa. Los sujetos fueron randomizados en una proporción de 1:1:1 dentro de 2 cohortes independientes (Cohorte 1/personas que no son de raza negra y Cohorte 2/personas de raza negra) y fueron estratificados según el genotipo del HCV (1a o 1b) y según la carga viral del ARN del HCV (menor que o igual a 400.000 IU por mL frente a más de 400.000 IU por mL) a uno de los siguientes 3 grupos de tratamiento: PegIntron + Rebetol durante 48 semanas (PR48). PegIntron + Rebetol durante 4 semanas seguidos por Victrelis de 800 mg 3 veces al día + PegIntron + Rebetol durante 24 semanas. Los participantes continuaron luego en diferentes regímenes con base en la terapia guiada por la respuesta (Victrelis-RGT) de la semana de tratamiento (ST) 8 hasta la ST24. Todos los participantes en este grupo de tratamiento tuvieron como límite 24 semanas de terapia con Victrelis. Los participantes con un ARN del VHC indetectable (Objetivo No Detectado) en la ST8 (aquellos que respondieron al tratamiento en forma temprana) y que también fueron negativos hasta la ST24 descontinuaron la terapia e ingresaron en el seguimiento en la visita de la ST28. Los participantes con un ARN del VHC detectable en la ST8 o en cualquier semana de tratamiento siguiente pero que fueron posteriormente negativos en la ST24 (aquellos que respondieron al tratamiento en forma tardía) se cambiaron en ciego al placebo en la visita de la ST28 y continuaron la terapia con PegIntron + Rebetol durante 20 semanas adicionales, para alcanzar una duración total del tratamiento de 48 semanas. PegIntron + Rebetol durante 4 semanas seguidos por Victrelis de 800 mg 3 veces al día + PegIntron + Rebetol durante 44 semanas (Victrelis-PR48), Todos los participantes con un ARN del VHC detectable en plasma en la ST24 fueron descontinuados del tratamiento. La Respuesta Virológica Sostenida (SVR) se definió como el ARN del VHC en plasma menor que 25 IU/mL en la semana de seguimiento 24. Los resultados de VHC-ARN en plasma en la semana de seguimiento 12 se utilizaron si no se contaba con los resultados de VHC-ARN en plasma en la semana de seguimiento 24. La edad media de los participantes randomizados fue de 49 ańos. La distribución racial de los participantes fue la siguiente: 82% blancos, 14% negros y 4% otros. La distribución de los participantes según el sexo fue 60% hombres y 40% mujeres. La adición de Victrelis a PegIntron y Rebetol aumentó significativamente las tasas de SVR en comparación con PegIntron y Rebetol solos en la cohorte combinada (del 63% al 66% en los grupos con Victrelis frente al 38% en el grupo control con PR48) en el caso de los participantes randomizados que recibieron por lo menos 1 dosis de cualquier fármaco del estudio (población del Grupo de Análisis Completo). Las tasas de SVR para las personas de raza negra que recibieron la combinación de Victrelis con PegIntron y Rebetol fueron del 42% al 53% en un análisis predefinido (véase la Tabla 10).
Ver Tabla
En los participantes con cirrosis en la basal, la respuesta virológica sostenida fue mayor en aquellos que recibieron tratamiento con la combinación de Victrelis con PegIntron y Rebetol durante 44 semanas después de una terapia de introducción con PegIntron y Rebetol (10/24, 42%) en comparación con aquellos que recibieron la RGT (5/16, 31%). Respuesta Virológica Sostenida (SVR) con base en los resultados de VHC-ARN en la ST8: La Tabla 11 presenta la respuesta virológica sostenida con base en los resultados de VHC-ARN en la ST8 en los pacientes que no recibieron tratamiento previo. El 57% (208/368) de los participantes en el grupo de VICTRELIS-RGT y el 56% (204/366) de los participantes del grupo de VICTRELIS-PR48 tuvieron un ARN del VHC indetectable (Objetivo no Detectado) en la ST8 (aquellos que respondieron al tratamiento en forma temprana) en comparación con el 17% de los participantes (60/363) del grupo PR48.
Ver Tabla
Entre los participantes con un ARN del VHC detectable en la ST8 que habían logrado un ARN del VHC indetectable(Objetivo no Detectado) en la ST24 y finalizaron por lo menos 28 semanas de tratamiento, las tasas de SVR fueron del 66% (45/68) el grupo de Victrelis-RGT (4 semanas de PegIntron y Rebetol, más 24 semanas de Victrelis con PegIntron y Rebetol, seguidas por 20 semanas de PegIntron y Rebetol solos) y del 75% (55/73) en el grupo de Victrelis-PR48 (4 semanas de PegIntron y Rebetol, seguidas de 44 semanas de Victrelis con PegIntron y Rebetol). Participantes con respuesta parcial y fracaso en una terapia anterior con peginterferón alfa y ribavirina: El estudio RESPOND-2 fue un estudio randomizado, de grupo paralelo, a doble ciego que comparó 2 regímenes terapéuticos de Victrelis de 800 mg por vía oral 3 veces al día en combinación con PR [PegIntron de 1.5 microgramos por kg a la semana por vía subcutánea y ribavirina basada en el peso (600-1400 mg al día por vía oral dividida en 2 veces al día)] con PR solo en participantes adultos que padecían de infección crónica por el virus de la hepatitis C (genotipo 1 del VHC) con sensibilidad demostrada a interferón (definida históricamente a través de una disminución en la carga viral de VHC-ARN mayor que o igual a 2 log10 en la semana 12, pero que nunca alcanzaron una SVR [aquellos que respondieron de manera parcial] o ARN del VHC indetectable al final del tratamiento previo con un posterior ARN del VHC detectable en plasma [aquellos que recayeron]). Los participantes con una disminución menor que 2 log10 en el ARN del VHC en la semana 12 del tratamiento previo (participantes que no respondieron al tratamiento previo) no fueron elegibles para el enrolamiento en esta prueba. Los participantes fueron randomizados en una proporción 1:2:2 y se les estratificó con base en la respuesta a su régimen calificador anterior (aquellos que recayeron frente a aquellos que respondieron de manera parcial) y según el subtipo del VHC (1a frente a 1b) a uno de los siguientes grupos de tratamiento: PegIntron + Rebetol durante 48 semanas (PR48). PegIntron + Rebetol durante 4 semanas seguidos por Victrelis de 800 mg 3 veces al día + PegIntron + Rebetol durante 32 semanas. Los participantes continuaron luego en diferentes regímenes de tratamiento con base en la terapia guiada por la respuesta (Victrelis-RGT) de la ST8 y la ST12. Todos los participantes en este grupo de tratamiento tuvieron como límite 32 semanas de tratamiento con Victrelis. Los participantes con un ARN del VHC indetectable (Objetivo no detectado) en la ST8 (aquellos que respondieron al tratamiento en forma temprana) y ST12 finalizaron la terapia en la visita de la ST36. Los participantes con un ARN del VHC detectable en la ST8 pero posteriormente indetectable (Objetivo no detectado) en la ST12 (aquellos que respondieron al tratamiento en forma tardía) se cambiaron en ciego al placebo en la visita de la ST36 y continuaron el tratamiento con PegIntron + Rebetol durante 12 semanas adicionales, para alcanzar una duración total del tratamiento de 48 semanas. PegIntron + Rebetol durante 4 semanas seguidos por Victrelis de 800 mg 3 veces al día + PegIntron + Rebetol durante 44 semanas (VICTRELIS-PR48). Todos los participantes con un ARN del VHC detectable en plasma en la ST12 fueron descontinuados del tratamiento. La respuesta virológica sostenida (SVR) se definió como el ARN del VHC en plasma menor a 25 IU/mL en la semana de seguimiento 24. Los resultados de VHC-ARN en plasma en la semana de seguimiento 12 se utilizaron si no se contaba con los resultados de VHC-ARN en plasma en la semana de seguimiento 24. La edad media de los participantes randomizados fue de 53 ańos. La distribución racial de los participantes fue la siguiente: 85% blancos, 12% negros y 3% otros. La distribución de los participantes según el sexo fue 67% hombres y 33% mujeres. La adición de Victrelis a la terapia con PegIntron y Rebetol aumentó significativamente las tasas de SVR en comparación con PegIntron/Rebetol solos (del 59% al 66% en los grupos con Victrelis frente al 23% del grupo control con PR48) en el caso de los participantes randomizados que recibieron por lo menos 1 dosis de cualquier fármaco del estudio (población del Grupo de Análisis Completo) (véase la Tabla 12).
Ver Tabla
En los participantes con cirrosis en la basal, la respuesta virológica sostenida fue mayor en aquellos que recibieron tratamiento con la combinación de Victrelis con PegIntron y Rebetol durante 44 semanas después de una terapia de introducción de 4 semanas con PegIntron y Rebetol (17/22, 77%) en comparación con aquellos que recibieron la RCT (6/17, 35%). Respuesta Virológica Sostenida (SVR) con base en los resultados de VHC-ARN en la ST8: La Tabla 13 presenta la respuesta virológica sostenida con base en los resultados de VHC-ARN en la ST8 en los participantes que han fracasado en una terapia anterior. El 46% (74/162) de los participantes el grupo de Victrelis-RGT y el 52% (84/161) de los participantes el grupo de Victrelis-PR48 tuvieron un ARN del VHC indetectable (Objetivo no detectado) en la ST8 (aquellos que respondieron al tratamiento en forma temprana) en comparación con el 9% (7/80) de los participantes el grupo PR48.
Ver Tabla
Entre los participantes con un ARN del VHC detectable en la ST8 que lograron un ARN del VHC indetectable (Objetivo no detectado) en la ST12 y finalizaron por lo menos 36 semanas de tratamiento, las tasas de SVR fueron del 79% (27/34) en el grupo de Victrelis-RGT (4 semanas de PegIntron y Rebetol, luego 32 semanas de Victrelis con PegIntron y Rebetol, seguidas por 12 semanas de PegIntron y Rebetol solos) y del 72% (29/40) el grupo de Victrelis-PR48 (4 semanas de pegIntron y rebetol, luego 44 semanas de Victrelis con pegIntron y rebetol). Sensibilidad a interferón durante la terapia de introducción con peginterferón alfa y ribavirina: Participantes que no recibieron tratamiento previo: En los participantes que no recibieron tratamiento previo evaluados en el estudio SPRINT-2, la sensibilidad a interferón (definida como una disminución mayor que o igual a 1 log10 en la carga viral en la ST4) fue un factor de predicción de una SVR. Los participantes tratados con Victrelis que demostraron sensibilidad a interferón en la ST4 alcanzaron tasas de SVR del 81% (203/252) en el grupo de Victrelis-RGT y del 79% (200/254) en el grupo de Victrelis-PR48, en comparación con el 52% (134/260) en los participantes tratados con PegIntron/Rebetol. Los sujetos tratados con Victrelis que demostraron escasa sensibilidad a interferón (definida como una disminución menor que 1 log10 en la carga viral en la ST4) alcanzaron tasas de SVR del 28% (27/97) en el grupo de Victrelis-RGT y del 38% (36/95) en el grupo de Victrelis-PR48, en comparación con el 4% (3/83) en los participantes tratados con PegIntron/Rebetol, Los participantes con una disminución menor que 0.5 log10 en la carga viral en la ST4 alcanzaron tasas de SVR del 28% (13/47) en el grupo de Victrelis-RGT y del 30% (11/37) en el grupo de Victrelis-PR48, en comparación con el 0% (0/25 en los participantes tratados con PegIntron/Rebetol. Se prevé que los participantes con una disminución menor que 0.5 log10 en la carga viral en la ST4 con la terapia con peginterferón alfa más ribavirina solos tendrán una respuesta nula (una disminución menor que 2 log10 en la carga viral en la ST12) al tratamiento con peginterferón alfa y ribavirina. Participantes con respuesta parcial y que fracasaron en una terapia anterior con interferón alfa y ribavirina: En los participantes que recayeron anteriormente y que respondieron de manera parcial evaluados en el estudio RESPOND-2, la sensibilidad a interferón (definida como una disminución mayor que o igual a 1 log10 en la carga viral en la ST4) fue un factor de predicción de una SVR. Los participantes tratados con Victrelis que demostraron sensibilidad a interferón en la ST4 alcanzaron tasas de SVR del 74% (81/110) en el grupo de Victrelis-TGR y del 79% (90/114) en el grupo de Victrelis-PR48, en comparación con el 27% (18/67) en los participantes tratados con PegIntron/Rebetol. Los participantes tratados con Victrelis que demostraron escasa sensibilidad a interferón (definida como una disminución menor que 1 log10 en la carga viral en la ST4) alcanzaron tasas de SVR del 33% (15/46) en el grupo de Victrelis-RGT y del 34% (15/44) en el grupo de Victrelis-PR48, en comparación con el 0% (0/12) en los participantes tratados con PegIntron/Rebetol. Pacientes que han fracasado la terapia previa: respondedores nulos previos con interferón y ribavirina: Provide es un estudio en curso, abierto, de una sola rama para Victrelis 800 mg por vía oral 3 veces al día en combinación PegIntron 1.5 mcg/kg/semana por vía subcutánea y basado en el peso de ribavirina (600-1.400 mg por día en dosis orales divididas en 2) en pacientes adultos con hepatitis C crónica (VHC) genotipo 1 que no lograron una SVR, mientras el grupo control de PegIntron/Rebetol, en los estudios Fase 2 y 3 de terapia combinada con Victrelis. Los sujetos que se inscribieron en Provide, 2 semanas después de la última dosis de PegIntron/Rebetol del estudio madre, recibieron Victrelis 800 mg 3 veces al día + PegIntron + Rebetol durante 44 semanas. Los sujetos que no pudieron inscribirse en este estudio dentro de 2 semanas recibieron PegIntron/Rebetol por 4 semanas, seguido de Victrelis 800 mg 3 veces al día + PegIntron + Rebetol durante 44 semanas. En aquellos sujetos que se encontraban con respuesta nula en el grupo control de PegIntron/Rebetol en el estudio madre, que recibieron PegIntron/Rebetol por 4 semanas siguiendo el tratamiento con Victrelis 800 mg 3 veces al día + PegIntron + Rebetol durante 44 semanas, el 38% (20/52) consiguieron la SVR, y la tasa de recaída fue del 14% (3/22). El uso de la reducción de dosis de ribavirina en comparación con el agente estimulante de la eritropoyesis (ESA) en el tratamiento de la anemia en sujetos no tratados previamente. Un estudio aleatorizado, de grupo paralelo de etiqueta abierta fue llevado a cabo para comparar 2 estrategias para el manejo de la anemia (uso de ESA versus la reducción de la dosis de ribavirina) en 687 sujetos con el CHC genotipo 1 infección no tratados previamente que se convirtieron en anémicos durante el tratamiento con 800 mg Victrelis por vía oral 3 veces al día junto con peginterferón alfa-2b 1.5 mg/kg/semana por vía subcutánea y basado en el peso de ribavirina (600-1.400 mg por vía oral 2 veces al día). El estudio incluyó a sujetos con concentraciones de hemoglobina en suero de menos de 15 g/dL. Los sujetos fueron tratados con 4 semanas de peginterferón alfa-2b y ribavirina seguidos por hasta 44 semanas de Victrelis junto con peginterferón alfa-2b y ribavirina. Si un sujeto se convirtió en anémico (hemoglobina sérica de aproximadamente · 10 g/dL en el período de tratamiento), el sujeto fue al azar en una proporción de 1:1, en el momento en que se convirtió en anémico, ya sea a la reducción de la dosis de ribavirina (N=249) o el uso de eritropoyetina (eritropoyetina 40.000 unidades por vía subcutánea 1 vez por semana) para la administración de la anemia (n=251). Si las concentraciones séricas de hemoglobina siguen disminuyendo hasta · 8.5 g/dL, los temas podrían ser tratados con intervenciones de la anemia adicionales, incluyendo la adición de eritropoyetina (18% de las personas en el grupo de reducción de la dosis de ribavirina) o la reducción de dosis de ribavirina (37% de ellos usan ESA en el grupo). La media de edad de los sujetos al azar fue de 49 ańos. La distribución por razas de los sujetos fue de la siguiente manera: 77% blancos, 19% negros, y otros 4%. La distribución de los temas de género por parte de los hombres era de 37% y 63% mujeres. La tasa general de SVR con intención de tratar de todos los sujetos enrolados (incluyendo aquellos sujetos que no fueron asignados al azar a la reducción de dosis de ribavirina o ESA para el tratamiento de la anemia) fue del 63% (431/687). La tasa de SVR en pacientes aleatorizados para recibir la reducción de la dosis de ribavirina fue de 71.5% (178/249), y la tasa de SVR en pacientes aleatorizados que recibieron ESA fue de 71% (178/251). La tasa de recaída en los pacientes aleatorizados que recibieron dosis reducidas de ribavirina fue del 10% (19/196), y la tasa de recaída en los pacientes aleatorizados con ESA fue del 9.6% (19/197).
|
|
Posología:
Victrelis se debe administrar en combinación con peginterferón alfa y ribavirina. La dosis de Victrelis es de 800 mg (4 cápsulas de 200 mg) 3 veces al día (cada 7 a 9 horas) con alimentos (una comida o refrigerio liviano) (véase la Tabla 1). La dosis máxima diaria es de 2.400 mg. En algunos subgrupos, las siguientes recomendaciones de administración de dosis difieren de la administración de dosis estudiada en las pruebas de Fase 3 (véase Estudios clínicos). Para la mayoría de las personas se recomienda una Terapia Guiada por la Respuesta (RGT), pero en los subgrupos elegidos como objetivo (por ejemplo, pacientes con cirrosis) se recomienda una administración de dosis más prolongada. Terapia combinada con Victrelis: Pacientes sin cirrosis que no recibieron tratamiento previo o que anteriormente fracasaron con una terapia con interferón y ribavirina. Inicie la terapia con peginterferón alfa y ribavirina durante 4 semanas (Semanas de Tratamiento 1-4). Ańada Victrelis de 800 mg (4 cápsulas de 200 mg) por vía oral 3 veces al día (cada 7 a 9 horas) al régimen de peginterferón alfa y ribavirina después de 4 semanas de tratamiento. Con base en los niveles del ARN del HCV del paciente en la semana de tratamiento (ST) 8, ST12 y ST24, utilice los siguientes lineamientos de la terapia guiada por la respuesta (RGT) para determinar la duración del tratamiento (véase la Tabla 1).
Ver Tabla
En caso de los pacientes que no recibieron tratamiento previo y que son poco sensibles a interferón (según se determinó en la ST4), se deberá considerar el tratamiento con 4 semanas de peginterferón alfa y ribavirina seguidas por 44 semanas de Victrelis de 800 mg por vía oral 3 veces al día (cada 7 a 9 horas) en combinación con peginterferón alfa y ribavirina, con la finalidad de maximizar las tasas de SVR [véase Estudios clínicos]. Terapia combinada con Victrelis/peginterferón alfa/ribavirina: Pacientes con cirrosis: Los pacientes con cirrosis compensada deberán recibir 4 semanas de peginterferón alfa y ribavirina seguidas por 44 semanas de Victrelis de 800 mg (4 cápsulas de 200 mg) 3 veces al día (cada 7 a 9 horas) en combinación con peginterferón alfa y ribavirina. Modificación de la dosis: No se recomienda reducir la dosis de Victrelis. Si un paciente tiene una reacción adversa seria posiblemente relacionada con peginterferón alfa y/o ribavirina, la dosis de peginterferón alfa y/o ribavirina debería ser reducida o descontinuada. Consulte el folleto de información al profesional de peginterferón alfa y ribavirina para obtener información adicional sobre cómo reducir y/o descontinuar la dosis de peginterferón alfa y/o ribavirina. Victrelis no se debe administrar en ausencia de peginterferón alfa y ribavirina. Si peginterferón o ribavirina es discontinuado permanentemente, Victrelis también debe ser discontinuado. Descontinuación de la administración de dosis debido a la ineficacia del tratamiento: Se recomienda descontinuar la terapia en todos los pacientes con 1) niveles del ARN del HCV mayores que o iguales a 100 IU por mL en la ST12 ó 2) niveles del ARN del HCV confirmados y detectables en la ST24.
|
|
Efectos Colaterales:
Véase los folletos de información al paciente de peginterferón alfa y ribavirina para obtener una descripción de las reacciones adversas asociadas con su uso. Experiencia en pruebas clínicas: Debido a que las pruebas clínicas se realizan en condiciones ampliamente variables, las tasas de reacciones adversas observadas en las pruebas clínicas de Victrelis no se pueden comparar directamente con las tasas de las pruebas clínicas de otro fármaco y probablemente no reflejen las tasas observadas en la práctica. Las siguientes reacciones adversas al fármaco (ADR) serias y de otra manera importantes se analizan detalladamente en otra sección del presente: Anemia (véase Advertencias e Información de orientación para el paciente). Neutropenia (véase Advertencias e Información de orientación para el paciente). Hipersensibilidad (véase Contraindicaciones, Advertencias, Precauciones e Información de orientación para el paciente). Las reacciones adversas informadas con mayor frecuencia (más del 35% de los participantes independientemente de la evaluación de la causalidad por parte del investigador) en participantes adultos fueron fatiga, anemia, náuseas, cefalea y disgeusia cuando Victrelis se utilizó en combinación con PegIntron/Rebetol. La seguridad de la combinación de Victrelis de 800 mg 3 veces al día con PegIntron/Rebetol se evaluó en 2095 participantes con hepatitis C crónica en una prueba de etiqueta abierta de Fase 2 y 2 pruebas clínicas de Fase 3 randomizadas, a doble ciego, controladas con placebo. El estudio SPRINT-1 (participantes que no recibieron tratamiento previo) evaluó el uso de Victrelis en combinación con PegIntron/Rebetol con o sin un período de introducción de 4 semanas con PegIntron/Rebetol en comparación con PegIntron/Rebetol solos. El estudio SPRINT-2 (sujetos que no recibieron tratamiento previo) y el estudio RESPOND-2 (sujetos que habían fracasado en una terapia anterior) evaluaron el uso de Victrelis de 800 mg 3 veces al día en combinación con PegIntron/Rebetol con un período de introducción de 4 semanas con PegIntron/Rebetol en comparación con PegIntron/Rebetol solos (véase Estudios clínicos). La población estudiada tenía una edad media de 49 ańos (3% de los participantes eran mayores que 65 ańos de edad), el 39% eran mujeres, el 82% eran blancos y el 15% eran negros. Durante el período de introducción de 4 semanas con PegIntron/Rebetol en los grupos con Victrelis, 28/1263 (2%) participantes presentaron reacciones adversas que condujeron a la descontinuación del tratamiento. Durante todo el curso del tratamiento, la proporción de los participantes que descontinuaron el tratamiento debido a reacciones adversas fue del 13% en el caso de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol y del 12% en el caso de los participantes que recibían PegIntron/Rebetol solos. Los eventos que resultaron en la descontinuación del tratamiento fueron similares a aquellos observados en estudios previos con PegIntron/Rebetol. Sólo la anemia y la fatiga se informaron como eventos que condujeron a la descontinuación en más del 1% de los participantes en cualquier grupo. Se produjeron reacciones adversas que condujeron a modificaciones de la dosis de cualquiera de los fármacos (principalmente PegIntron y Rebetol) en el 39% de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol en comparación con el 24% de los participantes que recibían PegIntron/Rebetol solos. La razón más común para la reducción de la dosis fue la anemia, que se presentó con más frecuencia en los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol que en los participantes que recibían PegIntron/Rebetol solos. Se informaron eventos adversos serios en el 11% de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol y en el 8% de los participantes que recibían PegIntron/Rebetol. Los eventos adversos (independientemente de la evaluación de la causalidad del investigador) informados en el 10% o más de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol, e informadosa una tasa del 5% o más que con PegIntron/Rebetol solos en los estudios SPRINT-1, SPRINT-2 y RESPOND-2 se presentan en la Tabla 3.
Ver Tabla
Otras importantes reacciones adversas informadas en las pruebas clínicas: Entre los participante(participantes que no recibieron tratamiento previo o aquellos que fracasaron en la terapia anterior) que recibieron Victrelis en combinación con peginterferón alfa y ribavirina, se informaron las siguientes reacciones adversas al fármaco. Estos eventos son notables debido a su seriedad, severidad o mayor frecuencia en los participantes que recibieron Victrelis en combinación con peginterferón alfa y ribavirina en comparación con los participantes que recibieron sólo peginterferón alfa y ribavirina. Trastornos gastrointestinales: La disgeusia (alteración del gusto) fue un evento adverso informado con una mayor frecuencia en los participantes que recibían Victrelis en combinación con peginterferón alfa y ribavirina en comparación con los participantes que recibían peginterferón alfa y ribavirina solos (Tabla 3). Eventos adversos como sequedad de boca, náuseas, vómitos y diarrea también se informaron con una mayor frecuencia en los participantes que recibían Victrelis en combinación con peginterferón alfa y ribavirina. Valores de laboratorio: En la Tabla 4 se describen los cambios en los parámetros hematológicos seleccionados durante el tratamiento de los participantes adultos con la combinación de Victrelis con PegIntron y Rebetol. Hemoglobina: Las disminuciones en la hemoglobina pueden requerir una disminución de la dosis/interrupción o descontinuación de ribavirina (véase Advertencias y Estudios clínicos; véase el Folleto de Información al Paciente de ribavirina). Si ribavirina es discontinuada permanentemente, también deben ser discontinuados peginterferón alfa y Victrelis (véase Posología). Neutrófilos y plaquetas: La proporción de los participantes con menores recuentos de neutrófilos y plaquetas fue mayor en los grupos con Victrelis en comparación que en los participantes que recibían PegIntron/Rebetol solos. El 3% de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol tuvo recuentos de plaquetas menores que 50 × 109 por L en comparación con el 1% de los participantes que recibían PegIntron/Rebetol solos. Las disminuciones de los neutrófilos o las plaquetas pueden requerir una reducción de la dosis o la interrupción de peginterferón alfa, o la descontinuación de la terapia (véase el Folleto de información al paciente de peginterferón alfa y ribavirina). Si peginterferón alfa es discontinuado permanentemente, también deben ser discontinuados ribavirina y Victrelis (véase Posología).
Ver Tabla
Experiencia postmarketing: Las siguientes experiencias adversas han sido notificadas durante la experiencia postcomercialización con el uso de Victrelis en combinación con peginterferón alfa y ribavirina. Debido a que estas reacciones son notificadas de forma voluntaria desde una población de tamańo incierto, no siempre es posible estimar una frecuencia de manera fiable o establecer una relación causal de la exposición al fármaco. Trastornos gastrointestinales: Ulceras bucales, estomatitis. Trastornos de la piel y tejido subcutáneo: Angioedema, urticaria (véase Advertencias); síndrome de erupción cutánea con eosinofilia y síntomas sistémicos (DRESS), erupción cutánea exfoliativa, dermatitis exfoliativa, síndrome de Stevens-Johnson, erupción cutánea tóxica, toxicodermia.
|
|
Contraindicaciones:
Las contraindicaciones para peginterferón alfa y ribavirina también se aplican al tratamiento combinado con Victrelis. El tratamiento de Victrelis con peginterferón alfa y ribavirina está contraindicado en: Mujeres embarazadas y hombres cuyas parejas de sexo femenino estén embarazadas debido a los riesgos de defectos congénitos y muerte fetal asociados con ribavirina (véase Advertencias, Precauciones y Uso en poblaciones específicas). Pacientes con historia de reacción de hipersensibilidad de boceprevir (véase Advertencias y Precauciones). Coadministración con fármacos que son altamente dependientes del CYP3A4/5 para su depuración, y para los cuales las concentraciones plasmáticas elevadas están asociadas con eventos serios y/o que ponen en riesgo la vida, tales como aquellos presentados en la Tabla 2, están contraindicadas (véase también Interacciones farmacológicas). Coadministración con potentes inductores del CYP3A4/5, en donde las concentraciones plasmáticas de boceprevir significativamente menores pueden estar asociadas con una menor eficacia, tales como aquellos presentados en la Tabla 2, están contraindicadas (véase también Interacciones farmacológicas).
Ver Tabla
|
|
Advertencias:
Anemia (uso con ribavirina y peginterferón alfa): Se ha reportado anemia con la terapia con peginterferón alfa y ribavirina. La adición de Victrelis a peginterferón alfa y ribavirina está asociada con una disminución adicional en las concentraciones de hemoglobina. Se deberán obtener recuentos sanguíneos completos antes del tratamiento y en las semanas de tratamiento 2, 4, 8 y 12, y se deberá realizar un monitoreo riguroso en otros puntos de evaluación, según sea clínicamente adecuado. Si la hemoglobina es menor que 10 g por dL, se recomienda una disminución de la dosis o la interrupción de ribavirina; y si la hemoglobina es menor que 8.5 g por dL, se recomienda la descontinuación de ribavirina (véase Efectos colaterales y Estudios clínicos). Si la discontinuación de ribavirina es requerida de forma permanente para manejar la anemia, también debe discontinuarse peginterferón alfa y Victrelis (véase Posología). Consulte el folleto de información al paciente de ribavirina para obtener información adicional con respecto a la reducción y/o interrupción de la dosis. En pruebas clínicas con Victrelis, la proporción de los participantes que presentaron valores de la hemoglobina menores que 10 g por dL y menores que 8.5 g por dL fue mayor en los participantes tratados con la combinación de Victrelis con PegIntron®/Rebetol® que en aquellos tratados con PegIntron/Rebetol solos (véase la Tabla 4). Con las intervenciones utilizadas para el tratamiento de la anemia en las pruebas clínicas, la disminución adicional promedio de la hemoglobina fue de aproximadamente 1 g por dL. En los ensayos clínicos, la mediana de tiempo hasta la aparición de la hemoglobina inferior a 10 g por dL a partir del inicio del tratamiento fue similar entre los pacientes tratados con la combinación Victrelis y PegIntron/Rebetol (71 días con un rango de 15 a 337 días), en comparación con los que recibieron PegIntron/Rebetol (71 días con un rango de 8-337 días). Ciertas reacciones adversas relacionadas con los síntomas de la anemia, tales como disnea, disnea de esfuerzo, mareos y síncope, se reportaron con más frecuencia en participantes que recibieron la combinación de Victrelis con PegIntron/Rebetol que en aquellos tratados con PegIntron/Rebetol solos (véase Efectos colaterales). En pruebas clínicas con Victrelis, las modificaciones de la dosis (generalmente de PegIntron/Rebetol) debido a la anemia se realizaron con el doble de frecuencia en los participantes tratados con la combinación de Victrelis con PegIntron/Rebetol (26%) en comparación con aquellos tratados con PegIntron/Rebetol (13%). La proporción de sujetos que descontinuaron el fármaco del estudio debido a la anemia fue del 1% en los participantes tratados con la combinación de Victrelis con PegIntron/Rebetol y del 1% en los participantes que recibieron PegIntron/Rebetol. Se permitió el uso de agentes estimulantes de la eritropoyesis para el tratamiento de la anemia, a criterio del investigador, con o sin la reducción de la dosis de ribavirina en las pruebas clínicas de Fase 2 y 3. La proporción de los participantes que recibieron un agente estimulante de la eritropoyesis fue del 43% en el grupo con Victrelis con PegIntron/Rebetol en comparación con el 24% de PegIntron/Rebetol solos. La proporción de sujetos que recibieron una transfusión para el tratamiento de la anemia fue del 3% de los participantes del grupo con Victrelis y PegIntron/Rebetol en comparación con menos del 1% en los participantes que recibieron PegIntron/Rebetol solos. Se han asociado eventos tromboembólicos con el uso de agentes estimulantes de la eritropoyesis en otras enfermedades, los cuales también se han informado con el uso de peginterferón alfa en los pacientes con hepatitis C. Se han informado eventos tromboembólicos en pruebas clínicas con Victrelis entre los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol y entre aquellos que recibían PegIntron/Rebetol solos, independientemente del uso de agentes estimulantes de la eritropoyesis. No se puede realizar una evaluación definitiva de la causalidad o una evaluación del riesgo-beneficio para estos eventos debido a la presencia de factores de confusión y la falta de randomización del uso de agentes estimulantes de la eritropoyesis. Un estudio aleatorizado, de grupo paralelo de etiqueta abierta fue llevado a cabo para comparar el uso de ESA versus la reducción de la dosis de ribavirina) en 687 sujetos con el CHC genotipo 1 infección no tratados previamente que se convirtieron en anémicos durante el tratamiento con 800 mg Victrelis por vía oral 3 veces al día junto con peginterferón alfa-2b 1.5 mg/kg/semana por vía subcutánea y basado en el peso de ribavirina (600-1.400 mg por vía oral 2 veces al día). Un estudio clínico aleatorio, de etiqueta abierta y de grupos paralelos, se llevó a cabo en pacientes con CHC no tratados previamente con infección genotipo 1 para comparar el uso de una reducción de la dosis de ribavirina en comparación con la ESA para el manejo inicial de la anemia durante el tratamiento con Victrelis en combinación con peginterferón alfa-2b y ribavirina. Las tasas de SVR reportadas fueron similares en pacientes que fueron asignados aleatoriamente para recibir la reducción de dosis de ribavirina en comparación con los sujetos que fueron asignados al azar para recibir un ESA. En este estudio, el uso de los ESA se asoció con un mayor riesgo de eventos tromboembólicos, incluyendo embolia pulmonar, infarto agudo de miocardio, accidente cerebrovascular y trombosis venosa profunda en comparación con la reducción de dosis de ribavirina sola. La tasa de interrupción del tratamiento debido a una anemia fue similar en los sujetos asignados al azar para recibir una reducción de dosis de ribavirina en comparación con sujetos asignados al azar para recibir la ESA (2% en cada grupo). La tasa de transfusión fue del 4% en los sujetos asignados al azar para recibir una reducción de dosis de ribavirina y el 2% en los sujetos asignados al azar para recibir la ESA. Se recomienda reducir la dosis de ribavirina para el tratamiento inicial de la anemia. Neutropenia (uso con ribavirina y peginterferón alfa): En estudios clínicos de Fase 2 y 3, el 7% de los participantes que recibían la combinación de Victrelis con PegIntron/Rebetol tuvo recuentos de neutrófilos menores que 0.5 × 109 por L en comparación con el 4% de los participantes que recibían PegIntron/Rebetol solos (véase la Tabla 4). 3 participantes presentaron infecciones severas o que ponen en riesgo la vida asociadas con neutropenia, y 2 participantes presentaron neutropenia que pone en riesgo la vida mientras recibían la combinación de Victrelis con PegIntron/Rebetol. Se debe realizar un hemograma completo (con recuentos diferenciales de leucocitos en todos los pacientes antes de iniciar la terapia combinada con Victrelis. Se deberán obtener hemogramas completos en las semanas de tratamiento 4, 8 y 12, y se deberá realizar un monitoreo riguroso en otros puntos de evaluación, según sea clínicamente adecuado. Las disminuciones en los recuentos de neutrófilos pueden requerir la reducción de la dosis o la descontinuación de peginterferón alfa y ribavirina. Si la discontinuación de peginterferón alfa y ribavirina es requerida, también debe discontinuarse Victrelis [véase Posología]. Consulte el folleto de información al paciente de peginterferón alfa y ribavirina para obtener información adicional con respecto a la reducción de la dosis o la descontinuación de peginterferón alfa y ribavirina. Hipersensibilidad: Reacciones graves y serias de hipersensibilidad aguda (por ejemplo urticaria, angioedema) se han observado durante el tratamiento combinado con Victrelis, peginterferón alfa, y ribavirina. Si la reacción se produce como tal, el tratamiento combinado se debe suspender y se debe iniciar inmediatamente la terapia médica apropiada (véase Contraindicaciones y Efectos colaterales). Interacciones farmacológicas: Véase la Tabla 2 para obtener un listado de los fármacos que están contraindicados para el uso con Victrelis debido a eventos adversos que posiblemente pongan en riesgo la vida, interacciones farmacológicas importantes o pérdida de la actividad virológica (véase Contraindicaciones). Consulte la Tabla 5 para obtener información sobre interacciones farmacológicas establecidas y otras posiblemente importantes (véase Interacciones farmacológicas). Pruebas de laboratorio: Los niveles del ARN del HCV se deberán monitorear en las semanas de tratamiento 4, 8, 12 y 24, al final del tratamiento, durante el seguimiento del tratamiento, y durante otros puntos de evaluación según se indique clínicamente. Se recomienda el uso de un ensayo sensible de reacción en cadena de la polimerasa de transcriptasa inversa (RT-PCR) en tiempo real para monitorear los niveles del ARN del HCV durante el tratamiento. El ensayo deberá tener un límite inferior de cuantificación del ARN del HCV igual o inferior a 25 IU por mL y un límite de detección del ARN del HCV de aproximadamente 10 a 15 IU por mL. Para los fines de evaluar los acontecimientos importantes de la terapia guiada por la respuesta, un resultado confirmado del ARN del HCV detectable pero por debajo del límite de cuantificación no se deberá considerar equivalente a un resultado del ARN del HCV indetectable (reportado como "Objetivo no detectado " o " VHC-ARN no detectado". Se debe realizar un hemograma completo (con recuentos diferenciales de leucocitos) en todos los pacientes antes de iniciar la terapia combinada con Victrelis. Se deberán obtener hemogramas completos en las semanas de tratamiento 2, 4, 8 y 12, y se deberá realizar un monitoreo riguroso en otros puntos de evaluación, según sea clínicamente adecuado. Consulte el Folleto de Información al Paciente de peginterferón alfa y ribavirina, incluidos los requisitos de las pruebas de embarazo.
|
|
Precauciones:
Embarazo (uso con ribavirina y peginterferón alfa): Ribavirina puede producir defectos congénitos y/o muerte del feto expuesto. Se debe tener extremo cuidado a fin de evitar el embarazo en las pacientes de sexo femenino y en las parejas de sexo femenino de los pacientes de sexo masculino. La terapia con ribavirina no se debería ser iniciada a menos que se haya obtenido en una prueba de embarazo resultado negativo inmediatamente antes del inicio de la terapia. Las mujeres potencialmente fértiles y los hombres deben utilizar por lo menos 2 métodos anticonceptivos eficaces durante el tratamiento y durante un mínimo de 6 meses después de finalizado el tratamiento. Una de esas formas de contracepción puede ser un anticonceptivo oral combinado que contenga al menos 1 mg de noretindrona. Anticonceptivos orales con bajas dosis de noretindrona y otras formas hormonas contraceptivas no han sido estudiadas o están contraindicadas. Durante este tiempo, se deben realizar pruebas de embarazo de rutina mensualmente (véase Contraindicaciones e Interacciones farmacológicas). Embarazo: Victrelis se debe administrar en combinación con peginterferón alfa y ribavirina (véase Posología). Categoría X para el Embarazo: Uso con Ribavirina y Peginterferón Alfa: Se han demostrado efectos teratogénicos y/o embriocidas significativos en todas las especies animales expuestas a ribavirina; por lo tanto, ribavirina está contraindicada en mujeres embarazadas y en las parejas de sexo masculino de mujeres embarazadas (véase Contraindicaciones, Advertencias y Folletos de Información al Paciente de ribavirina). Los interferones tienen efectos abortivos en los animales y se deberá suponer que tienen potencial abortivo en los seres humanos (véase Folleto de Información al Paciente de Peginterferón alfa). Se debe tener extremo cuidado a fin de evitar el embarazo en las pacientes de sexo femenino y en las parejas de sexo femenino de los pacientes de sexo masculino mientras toman esta combinación. Las mujeres potencialmente fértiles y sus parejas de sexo masculino no deberán recibir ribavirina a menos que utilicen métodos anticonceptivos eficaces (2 métodos confiables) durante el tratamiento con ribavirina y durante los 6 meses posteriores al tratamiento. Una de esas formas de contracepción puede ser un anticonceptivo oral combinado que contenga al menos 1 mg de noretindrona. Anticonceptivos orales con bajas dosis de noretindrona y otras formas hormonas contraceptivas no han sido estudiadas o están contraindicadas (véase Contraindicaciones y Advertencias). Categoría B para el Embarazo: Victrelis: Victrelis no se debe utilizar como monoterapia. No existen estudios adecuados ni bien controlados con Victrelis en mujeres embarazadas. No se han observado efectos sobre el desarrollo fetal en ratas y conejos a exposiciones del área bajo la curva (AUC) de boceprevir de aproximadamente 11.8 y 2.0 veces mayores, respectivamente, que aquellas en seres humanos a la dosis recomendada de 800 mg 3 veces al día (véase Toxicología no clínica). Madres que dan de lactar: Se desconoce si Victrelis se excreta en la leche materna humana. Los niveles de boceprevir y/o los metabolitos en la leche de ratas que dan de lactar fueron ligeramente mayores que los niveles observados en la sangre materna. Las concentraciones sanguíneas máximas de boceprevir y/o los metabolitos en las crías lactantes fueron menores que el 1% de las concentraciones sanguíneas maternas. Debido al potencial de reacciones adversas originadas por el fármaco en bebés lactantes, se debe decidir si se descontinúa la lactancia o se descontinúa el tratamiento con Victrelis, tomando en cuenta la importancia de la terapia para la madre. Uso pediátrico: No se ha estudiado la seguridad, la eficacia ni el perfil farmacocinético de Victrelis en pacientes pediátricos. Uso geriátrico: Los estudios clínicos de Victrelis no incluyeron un número suficiente de participantes de 65 ańos de edad y mayores para determinar si responden de manera diferente de los participantes más jóvenes. En general, se deberá tener cuidado en la administración y el monitoreo de Victrelis en los pacientes geriátricos debido a una mayor frecuencia de función hepática reducida, enfermedades concomitantes y otras terapias farmacológicas (véase Farmacología clínica). Insuficiencia renal: No se requiere el ajuste de la dosis de Victrelis en pacientes con cualquier grado de insuficiencia renal (véase Farmacología clínica). Insuficiencia hepática: No se requiere el ajuste de la dosis de Victrelis en pacientes con insuficiencia hepática leve, moderada o severa (véase Farmacología clínica). No se ha estudiado la seguridad y la eficacia de Victrelis en pacientes con cirrosis descompensada. Véase el Folleto de información al paciente de peginterferón alfa para obtener información sobre las contraindicaciones en la descompensación hepática. Coinfección por el virus de inmunodeficiencia humana (VIH): No se ha establecido la seguridad ni la eficacia de Victrelis sólo o en combinación con peginterferón alfa y ribavirina para el tratamiento de la infección crónica por el virus de la hepatitis C de genotipo 1 en pacientes coinfectados por VIH y HCV. Para obtener información referente a las interacciones medicamentosas con agentes antirretrovirales en participantes sanos (véase Interacciones farmacológicas y Farmacología clínica). Coinfección por el virus de la hepatitis B (HBV): No se ha estudiado la seguridad ni la eficacia de Victrelis sólo o en combinación con peginterferón alfa y ribavirina para el tratamiento de la infección crónica por el virus de la hepatitis C de genotipo 1 en pacientes coinfectados por HBV y HCV. Trasplante de órganos: No se ha estudiado la seguridad ni la eficacia de Victrelis sólo o en combinación con peginterferón alfa y ribavirina para el tratamiento de la infección crónica por el virus de la hepatitis C de genotipo 1 en receptores de trasplante de hígado u otro órgano. Carcinogénesis, mutagénesis, trastorno de la fertilidad: Carcinogénesis y mutagénesis: Uso con ribavirina y peginterferón alfa: Ribavirina es genotóxica en ensayos in vitro e in vivo. Ribavirina no fue oncogénica en estudios de carcinogenicidad realizados en ratones y ratas a dosis menores que la dosis diaria máxima recomendada en seres humanos. Consulte el folleto de información al paciente de ribavirina para obtener información adicional. Se llevaron a cabo estudios de carcinogenicidad de dos ańos de duración con boceprevir en ratones y ratas. Los ratones macho recibieron dosis de hasta 500 mg por kg y los ratones hembra recibieron dosis de hasta 650 mg por kg. Por otra parte, las ratas macho recibieron dosis de hasta 125 mg por kg y las ratas hembras recibieron dosis de hasta 100 mg por kg. En los ratones, no se observaron aumentos significativos en la incidencia de neoplasias relacionadas con el fármaco a las dosis máximas sometidas a prueba que produjeron exposiciones del AUC de boceprevir aproximadamente 2.3 y 6.0 veces mayores en los machos y las hembras, respectivamente, que aquellas en los seres humanos a la dosis recomendada de 800 mg 3 veces al día. En las ratas, no se observaron aumentos en la incidencia de neoplasias relacionadas con el fármaco a las dosis máximas sometidas a prueba que produjeron exposiciones del AUC de boceprevir similares a aquellas en los seres humanos a la dosis recomendada de 800 mg 3 veces al día. Boceprevir no fue genotóxico en una serie de ensayos in vitro o in vivo, tales como ensayos de mutagenicidad bacteriana, de aberración cromosómica en linfocitos de sangre periférica humana y en micronúcleo de ratón. Trastorno de la fertilidad: Uso con ribavirina y peginterferón alfa: En estudios de fertilidad realizados en animales machos, ribavirina indujo la toxicidad testicular reversible, mientras que peginterferón alfa puede deteriorar la fertilidad en las hembras. Consulte los folletos de información al paciente de ribavirina y peginterferón alfa para obtener información adicional. Boceprevir indujo efectos reversibles en la fertilidad y el desarrollo embrionario temprano en las ratas hembra, y no se observaron efectos a un nivel de dosis de 75 mg por kg. A esta dosis, las exposiciones del AUC de boceprevir son aproximadamente 1,3 veces mayores que aquellas en los seres humanos a la dosis recomendada de 800 mg 3 veces al día. También se observó una menor fertilidad en las ratas macho, más bien como una consecuencia de la degeneración testicular. No se observó degeneración testicular a un nivel de dosis de 15 mg por kg que produjo exposiciones del AUC de boceprevir menores que aquellas en los seres humanos a la dosis recomendada de 800 mgs 3 veces al día. No se observó degeneración testicular en los ratones o monos que recibieron boceprevir durante 3 meses a dosis de hasta 900 o 1000 mg por kg, respectivamente. A estas dosis, las exposiciones del AUC de boceprevir son aproximadamente 6.8 y 4.4 veces mayores en los ratones y monos, respectivamente, que aquellas en los seres humanos a la dosis recomendada de 800 mg 3 veces al día. Adicionalmente, el monitoreo clínico limitado no ha revelado evidencia de toxicidad testicular en los seres humanos.
|
|
Interacciones Medicamentosas:
Véase también Contraindicaciones, Advertencias y Farmacología clínica. Posibilidad de que Victrelis afecte a otros fármacos: Boceprevir es un fuerte inhibidor del CYP3A4/5. Los fármacos metabolizados principalmente por el CYP3A4/5 pueden tener una exposición mayor cuando se administran con Victrelis, que podría aumentar o prolongar sus efectos terapéuticos y adversos. Boceprevir no inhibe al CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP2E1 in vitro. Además, boceprevir no induce al CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 o CYP3A4/5 in vitro. Boceprevir es un posible inhibidor de la glucoproteína P (P-gp) con base en los estudios in vitro. En un ensayo de interacción de medicamentos conducido con digoxina, Victrelis había limitado el potencial inhibidor de la glucoproteína P a concentraciones clínicamente relevantes. Posibilidad de que otros fármacos afecten a Victrelis: Boceprevir es metabolizado principalmente por la aldocetorreductasa (AKR). En pruebas de interacción farmacológica llevadas a cabo con inhibidores de la AKR diflunisal e ibuprofeno, la exposición a boceprevir no aumentó en una medida clínicamente significativa. Victrelis puede coadministrarse con los inhibidores de la AKR. Boceprevir es metabolizado parcialmente por el CYP3A4/5. También es un sustrato de la glucoproteína P. La coadministración de Victrelis con fármacos que inducen o inhiben el CYP3A4/5 podría disminuir o aumentar la exposición a boceprevir. Interacciones farmacológicas establecidas y otras posiblemente importantes: A continuación, se proporcionan recomendaciones con base en las interacciones farmacológicas establecidas o posiblemente importantes desde el punto de vista clínico. Victrelis está contraindicado con fármacos que son potentes inductores del CYP3A4/5 y fármacos que son altamente dependientes del CYP3A4/5 para su depuración, y para los cuales las concentraciones plasmáticas elevadas están asociadas con eventos serios y/o que ponen en riesgo la vida (véase Contraindicaciones). Interacciones farmacológicas establecidas y otras posiblemente importantes: Clase de fármaco concomitante: Nombre del fármaco. Efecto en la concentración de boceprevir o fármaco concomitante. Recomendaciones. Antiarrítmicos: amiodarona, bepridil, propafenona, quinidina. Antiarrítmicos: La coadministración con Victrelis tiene el potencial de producir eventos adversos serios y/o que ponen en riesgo la vida y no se ha estudiado. Es necesario proceder con precaución y se recomienda el monitoreo de la concentración terapéutica de estos fármacos si se utilizan en forma concomitante con Victrelis. Digoxina. Digoxina: Las concentraciones de digoxina pueden aumentar con Victrelis (véase Farmacología clínica). Medir las concentraciones séricas de digoxina antes de iniciar Victrelis. Continuar monitoreando las concentraciones de digoxina; consulte el folleto de información al profesional de digoxina respecto a la información de titulación de la dosis de digoxina. Anticoagulantes: warfarina. Warfarina: Las concentraciones de warfarina pueden alterarse cuando se coadministra con Victrelis. Monitoree rigurosamente el INR. Antidepresivos: trazodona, desipramina. Trazodona; desipramina. Las concentraciones plasmáticas de trazodona y desipramina pueden aumentar cuando se administran con Victrelis, lo cual origina eventos adversos como mareos, hipotensión y síncope. Utilice con precaución y considere una dosis menor de trazodona o desipramina. Escitalopram: Escitalopram: La exposición de escitalopram se redujo ligeramente cuando se coadministra con Victrelis. Los inhibidores selectivos de la recaptación de serotonina tales como escitalopram tienen un índice terapéutico amplio, sin embargo, puede ser necesario ajustar la dosis cuando se combina con Victrelis. Antifúngicos: ketoconazol*, itraconazol, posaconazol, voriconazol: Boceprevir; itraconazol; ketoconazol; posaconazol; voriconazol. Las concentraciones plasmáticas de ketoconazol, itraconazol, voriconazol o posaconazol pueden aumentar con Victrelis. Cuando se requiere la coadministración, las dosis de ketoconazol e itraconazol no deben exceder los 200 mg/día. Antigotosos: colchicina. Colchicina. Se prevé aumentos significativos en los niveles de colchicina; se ha informado toxicidad mortal de la colchicina con otros fuertes inhibidores del CYP3A4. No se deberá administrar colchicina con Victrelis a los pacientes con insuficiencia renal o hepática. Tratamiento de los ataques de gota (durante el tratamiento con Victrelis): 0.6 mg (1 comprimido) × 1 dosis, seguida por 0.3 mg (1/2 comprimido) 1 hora más tarde. La dosis se debe repetir no antes de los 3 días. Profilaxis de los ataques de gota (durante el tratamiento con Victrelis): Si el régimen original era de 0,6 mg dos veces al día, reduzca la dosis a 0.3 mg una vez al día. Si el régimen original era de 0.6 mg 1 vez al día, reduzca la dosis a 0.3 mg 1 vez cada 2 días. Tratamiento de la fiebre mediterránea familiar (FMF) (durante el tratamiento con Victrelis): Dosis diaria máxima de 0.6 mg (puede administrarse a una dosis de 0.3 mg 2 veces al día). Antiinfecciosos: claritromicina. Claritromicina. Las concentraciones de claritromicina pueden aumentar con Victrelis; sin embargo, no es necesario el ajuste de la dosis en los pacientes con función renal normal. Antimicobacterianos: rifabutina: Boceprevir; rifabutina: Se prevén aumentos en la exposición de rifabutina, mientras que la exposición de boceprevir puede disminuir. No se han establecido dosis para los 2 fármacos cuando se utilizan en forma combinada. No se recomienda el uso concomitante. Bloqueadores de los canales de calcio tipo dihidropiridina: felodipino, nifedipina, nicardipina. Dihidropiridina; bloqueadores de canales de calcio. Las concentraciones plasmáticas de los bloqueadores de canales de calcio tipo dihidropiridina pueden aumentar cuando se administran con Victrelis. Es necesario proceder con precaución y se recomienda el monitoreo clínico. Corticosteroides sistémicos: dexametasona. Boceprevir. La coadministración de Victrelis con inductores del CYP3A4/5 puede disminuir las concentraciones plasmáticas de boceprevir, lo cual puede provocar la pérdida del efecto terapéutico. Por lo tanto, esta combinación se deberá evitar de ser posible, y utilizar con precaución de ser necesario. Prednisona*. Prednisona. Las concentraciones de prednisona y su metabolito activo, prednisolona, aumenta cuando se administra con Victrelis (véase Farmacología clínica). No es necesario ajuste de dosis de prednisona durante su coadministración con Victrelis. Los pacientes que reciben prednisona y Victrelis deberían ser estrechamente monitoreados. Corticosteroides inhalados: budesonida, fluticasona. Budesonida; fluticasona. El uso concomitante de budesónida o fluticasona inhaladas con Victrelis puede originar el aumento de las concentraciones plasmáticas de budesonida o fluticasona, lo cual da como resultado concentraciones séricas de cortisol significativamente menores. De ser posible, evite la coadministración especialmente durante períodos prolongados. Antagonistas de los receptores de la endotelina: bosentán. Bosentán. Las concentraciones de bosentán pueden aumentar cuando se coadministra con Victrelis. Utilice con precaución y monitoree rigurosamente. Inhibidor de la Integrasa del VIH: Raltegravir*. raltegravir. No se requiere ajuste de dosis para Victrelis o Raltegravir. Inhibidores no nucleósidos de la transcriptasa inversa del VIH: efavirenz*. Boceprevir. Las concentraciones plasmáticas mínimas de boceprevir disminuyeron cuando Victrelis se coadministró con efavirenz, lo cual puede provocar la pérdida del efecto terapéutico. Evite la combinación. Etravirina*. Etravirina. Disminución de las concentraciones de etravirina co-administrado con Victrelis. La significancia clínica de las reducciones en los parámetros farmacocinéticos de etravirina no ha sido evaluada directamente. Inhibidores de la proteasa del VIH: atazanavir/ritonavir*. Atazanavir; ritonavir. La administración concomitante de boceprevir y atazanavir/ritonavir dio como resultado menores exposiciones en equilibrio dinámico a atazanavir y ritonavir. No se recomienda la coadministración de atazanavir/ritonavir y boceprevir. Darunavir/ritonavir*. Darunavir; ritonavir; boceprevir. La administración concomitante de boceprevir y darunavir/ritonavir dio como resultado menores exposiciones en equilibrio dinámico a boceprevir, darunavir y ritonavir. No se recomienda la coadministración de darunavir/ritonavir y boceprevir. Lopinavir/ritonavir*. Lopinavir; ritonavir; boceprevir. La administración concomitante de boceprevir y lopinavir/ritonavir dio como resultado menores exposiciones en equilibrio dinámico a boceprevir, lopinavir y ritonavir. No se recomienda la coadministración de lopinavir/ritonavir y boceprevir. Ritonavir*. Boceprevir. Cuando boceprevir se administra con ritonavir sólo, las concentraciones de boceprevir disminuyen. Inhibidores de la HMG-CoA Reductasa: atorvastatina*. Atorvastatina. La exposición a atorvastatina se aumenta cuando se administra con Victrelia. Utilice la dosis eficaz más baja de atorvastatina, pero no se exceda la dosis diaria de 40 mg cuando se administra junto con Victrelis. Pravastatina*: Pravastatina. La administración concomitante de pravastatina con Victrelis incrementa la mayor exposición a pravastatina. El tratamiento con pravastatina puede iniciarse con la dosis recomendada cuando se coadministra con Victrelis. Se justifica un estricto control clínico. Inmunosupresores: Ciclosporina*; tacrolimus*; sirolimus. ciclosporina; tracolimus; sirolimus. Los ajustes de dosis de ciclosporina deben preverse cuando se administra con Victrelis y debe ser guiado por un estrecho seguimiento de las concentraciones sanguíneas de ciclosporina y evaluaciones frecuentes de la función renal y de los efectos secundarios relacionados con ciclosporina. La administración concomitante de Victrelis con tracrolimus requiere reducción de la dosis y la prolongación significativa del intervalo de dosis de tacrolimus, con una estrecha monitorización de las concentraciones de tacrolimus en sangre y evaluaciones frecuentes de la función renal y de los efectos secundarios relacionados con tacrolimus. Las concentraciones sanguíneas de sirolimus se espera que aumenten significativamente cuando se administra con Victrelis. Se recomienda una estrecha monitorización de los niveles sanguíneos de sirolimus. Beta-agonistas inhalados: salmeterol. Salmeterol. No se recomienda el uso concurrente de salmeterol inhalado y Victrelis debido al riesgo de eventos cardiovasculares asociados con salmeterol. Analgésicos narcóticos/dependencia de opioides: metadona*. R-metadona. Las concentraciones plasmáticas de R-metadona disminuyen cuando se coadministran con Victrelis (véase Farmacología clínica). Los cambios observados no fueron de significancia clínica relevante. No se recomienda ajuste de dosis de Victrelis o metadona. Los pacientes pueden requerir un ajuste adicional de la dosis de metadona cuando se inicia o se detiene Vicrtelis para asegurar el efecto clínico de la metadona. Buprenorfina/naloxona*. Buprenorfina/naloxona. Las concentraciones plasmática de buprenorfina y naloxona aumentaron cuando se coadministra con Victrelis (véase Farmacología clínica). Los cambios observados no fueron de significancia clínica relevante. No se recomienda ajuste de dosis de Victrelis o buprenorfina/naloxona. Anticonceptivos hormonales orales: drospirenona/etinilestradiol*. Drospirenona. Las concentraciones de drospirenona aumentaron en presencia de boceprevir. Por lo tanto, el uso de productos que contiene drospirenona está contraindicado durante el tratamiento con Victrelis debido a los potenciales efectos de hiperkalemia (véase Contraindicaciones). Noretindrona/etinilestradiol*. Etinilestradiol; noretindrona. Las concentraciones de etinilestradiol disminuyeron en presencia de boceprevir. La Cmax de noretindrona disminuyó 17% en presencia de boceprevir (véase Farmacología clínica). La coadministración de Victrelis con un anticonceptivo combinado oral que contenga etinilestradiol y al menos 1 mg de noretindrona es poco probable que altere la efectividad de este anticonceptivo combinado oral (véase Uso en poblaciones específicas). Las pacientes que usan estrógenos como terapia de reemplazo hormonal debería ser monitoriadas clínicamente en lo que respecta a los síntomas de la deficiencia estrogénica. Inhibidores de la PDE5: sildenafilo, tadalafilo, vardenafilo. Sildenafilo; tadalafilo; vardenafilo. Se prevén aumentos en las concentraciones de los inhibidores de la PDE5, los cuales pueden ocasionar un aumento en los eventos adversos, que incluyen hipotensión, síncope, alteraciones visuales y priapismo. El uso de sildenafil o tadalafil para el tratamiento de la hipertensión arterial pulmonar (PAH) está contraindicado con Victrelis (véase Contraindicaciones). Uso de inhibidores de la PDE5 para la disfunción eréctil: Utilícelos con precaución en combinación con Victrelis, con un monitoreo más riguroso para detectar eventos adversos asociados con los inhibidores de la PDE5. No exceda las siguientes dosis: Sildenafil: 25 mg cada 48 horas. Tadalafil: 10 mg cada 72 horas. Vardenafil: 2.5 mg cada 24 horas. Inhibidor de la bomba de protones: omeprazol*. Omeprazol. Es recomendado no ajustar la dosis de omeprazol o Victrelis. Sedantes/hipnóticos: Alprazolam; midazolam I.V. Midazolam; alprazolam. Se deberá realizar un monitoreo clínico riguroso de la depresión respiratoria y/o la sedación prolongada durante la coadministración con Victreli. Se deberá considerar una dosis IV menor de midazolam o alprazolam. *Estas combinaciones se han estudiado; véase Farmacología clínica para obtener información sobre la magnitud de la interacción.
|
|
Sobredosificación:
Voluntarios sanos han tomado dosis diarias de 3.600 mg durante 5 días sin efectos sintomáticos perjudiciales. No existe un antídoto específico para la sobredosis con Victrelis. El tratamiento de la sobredosis con Victrelis deberá comprender medidas complementarias generales, tales como el monitoreo de los signos vitales y la observación del estado clínico del paciente.
|
|
Conservación:
Almacenamiento y manipulación: Las cápsulas de Victrelis deberán estar refrigeradas a una temperatura de 2-8°C hasta su administración. Evite la exposición al calor excesivo. Para uso del paciente, las cápsulas refrigeradas de Victrelis pueden permanecer estables hasta la fecha de expira impresa en la etiqueta. Victrelis también se puede almacenar a temperatura ambiente hasta 30°C por 3 meses, durante su uso. Mantenga el envase bien cerrado.
|
|
Observaciones:
Información de orientación para el paciente: Véase el Folleto de Información al Paciente aprobada por la FDA (Guía del Fármaco): Victrelis debe utilizarse en combinación con peginterferón alfa y ribavirina; por ende, también se aplican todas las contraindicaciones y advertencias correspondientes a peginterferón alfa y ribavirina. Embarazo: Ribavirina no se debe utilizar en mujeres que estén embarazadas ni en hombres cuyas parejas de sexo femenino estén embarazadas. No se deberá iniciar la terapia con ribavirina hasta que se haya obtenido un informe de una prueba de embarazo con resultado negativo inmediatamente antes del inicio de la terapia. Se debe informar a las pacientes de sexo femenino potencialmente fértiles y a los pacientes de sexo masculino con parejas de sexo femenino potencialmente fértiles sobre los riesgos teratogénicos/embriocidas de ribavirina, y se les debe indicar el uso de un método anticonceptivo eficaz durante la terapia y durante los 6 meses posteriores a la terapia. Se deberá recomendar a los pacientes que notifiquen inmediatamente a su profesional sanitario en caso de un embarazo (véase Contraindicaciones, Advertencias y Precauciones). Las mujeres potencialmente fértiles y los hombres deben utilizar por lo menos dos métodos anticonceptivos eficaces durante el tratamiento y durante un mínimo de 6 meses después de finalizado el tratamiento. Durante este tiempo, se deben realizar pruebas de embarazo de rutina mensualmente. Una de esas formas de contracepción puede ser un anticonceptivo oral combinado que contenga al menos 1 mg de noretindrona. Anticonceptivos orales con bajas dosis de noretindrona y otras formas hormonas contraceptivas no han sido estudiadas o están contraindicadas (véase Contraindicaciones, Advertencias y Precauciones). Anemia: Se deberá informar a los pacientes que la anemia puede aumentar cuando Victrelis se administra con peginterferón alfa y ribavirina (véase Advertencias, Precauciones y Efectos colaterales). Se deberá informar a los pacientes que se requieren evaluaciones de laboratorio antes de iniciar la terapia y periódicamente a partir de entonces (véase Advertencias y Precauciones). Neutropenia: Se deberá informar a los pacientes que la neutropenia puede aumentar cuando Victrelis se administra con peginterferón alfa y ribavirina (véase Advertencias, Precauciones y Efectos colaterales). Se deberá informar a los pacientes que se requieren evaluaciones de laboratorio antes de iniciar la terapia y periódicamente a partir de entonces (véase Advertencias y Precauciones). Hipersensibilidad: Reacciones graves y serias de hipersensibilidad aguda (por ejemplo urticaria, angioedema) se han observado durante el tratamiento combinado con Victrelis, peginterferón alfa, y ribavirina (véase Contraindicaciones, Advertencias y Precauciones). Si los síntomas de las reacciones hipersensibilidad aguda (por ejemplo, picor, urticaria, hinchazón de la cara, ojos, labios, lengua o garganta, problemas para respirar o tragar) se producen, los pacientes deben acudir al médico sin demora. Medidas preventivas sobre el uso: Se deberá recomendar a los pacientes que Victrelis no se debe utilizar solo debido a la alta probabilidad de resistencia sin las terapias combinadas anti-HCV (véase Indicaciones y Uso). Véase los Folletos de Información al Profesional de peginterferón alfa y ribavirina para obtener información de orientación adicional para el paciente sobre el uso de estos fármacos en combinación con Victrelis. Se deberá informar a los pacientes acerca del potencial de interacciones farmacológicas serias con Victrelis y de que algunos fármacos no se deberán tomar con Victrelis (véase Contraindicaciones, Advertencias, Precauciones, Interacciones farmacológicas y Farmacología clínica). Se deberá informar a los pacientes que la dosis total diaria de Victrelis está empacada en un frasco único que contiene 12 cápsulas y que el paciente deberá tomar 4 cápsulas 3 veces al día con los alimentos. Dosis perdidas de Victrelis: Si un paciente obvia una dosis y faltan menos de 2 horas para la siguiente dosis, se deberá omitir la dosis perdida. Si un paciente obvia una dosis y faltan 2 horas o más para la siguiente dosis, el paciente deberá tomar la dosis perdida con alimentos y reanudar el programa de administración de dosis habitual. Transmisión del virus de la hepatitis C: Se deberá informar a los pacientes que se desconoce el efecto del tratamiento de la infección por hepatitis C en la transmisión y que se deberán tomar las precauciones pertinentes para evitar la transmisión del virus de la hepatitis C.
|
|
Presentaciones:
Envase conteniendo 336 cápsulas.
|
|



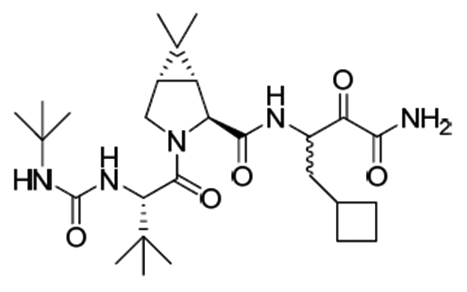{kind=link}
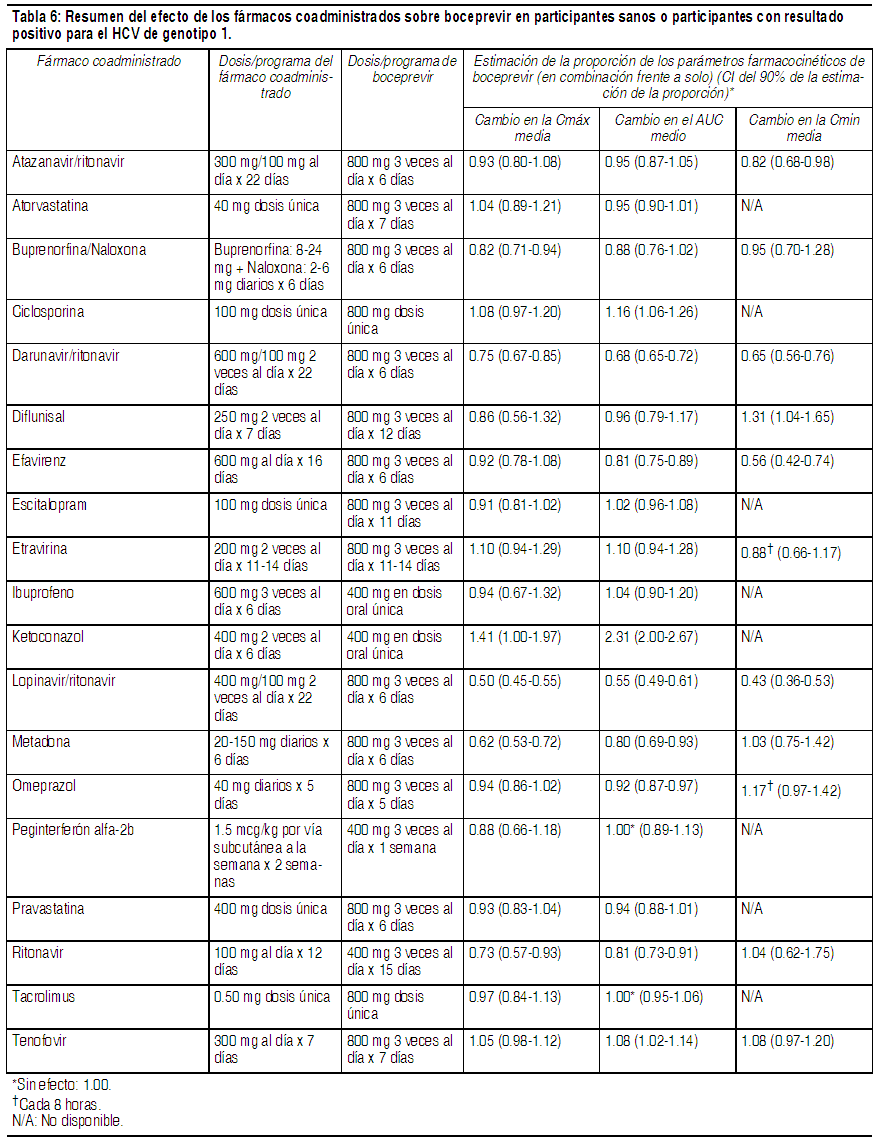{kind=link}
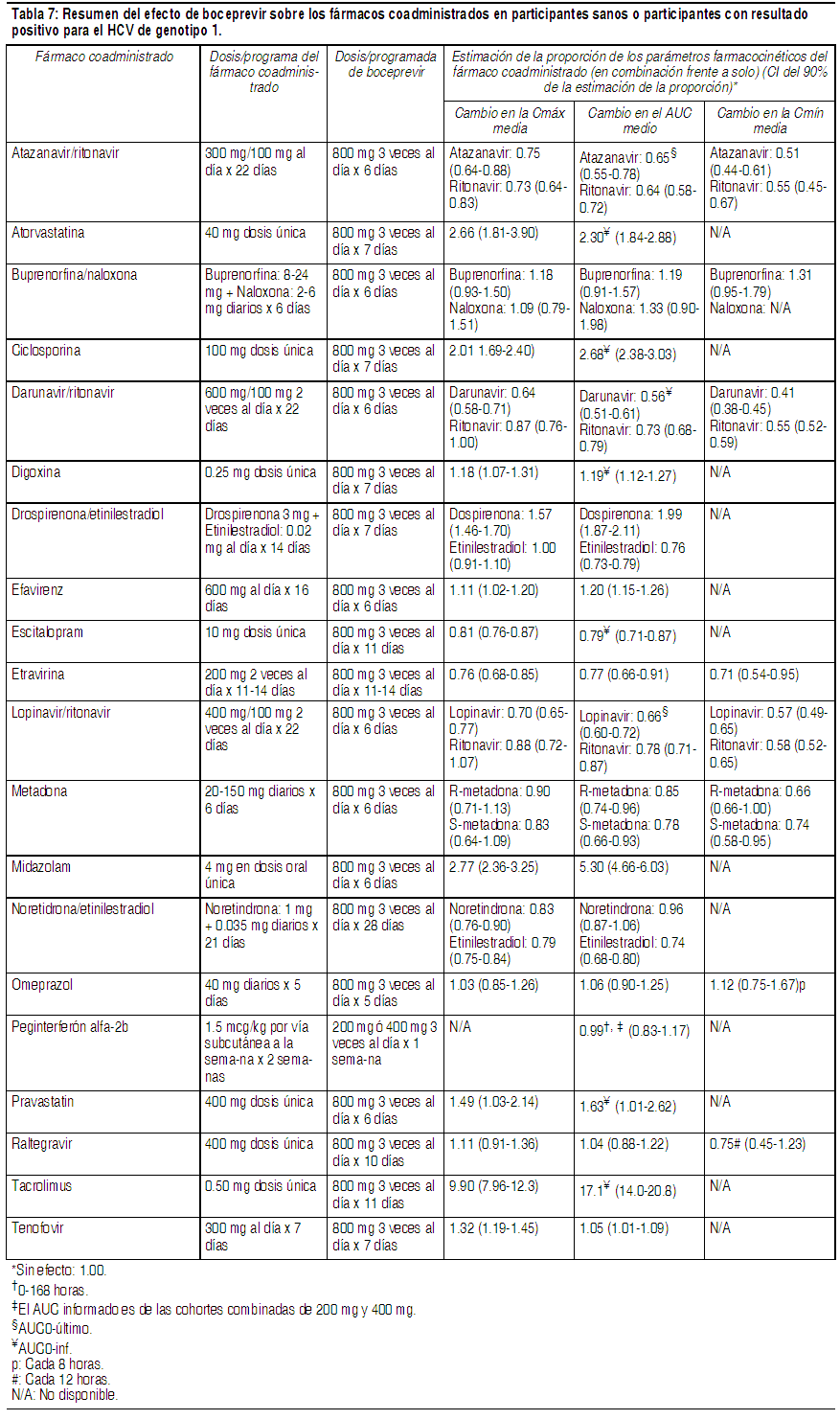{kind=link}
{kind=link}
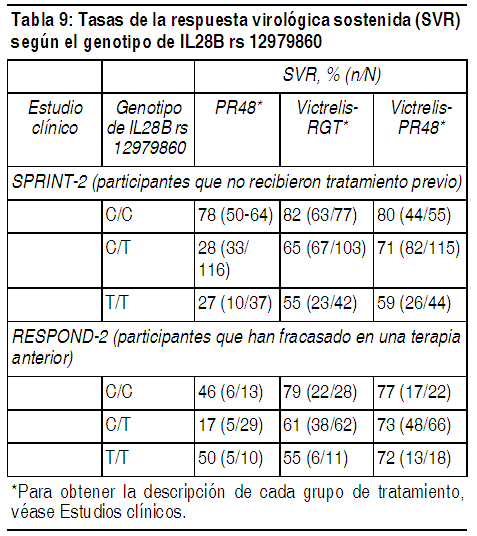{kind=link}
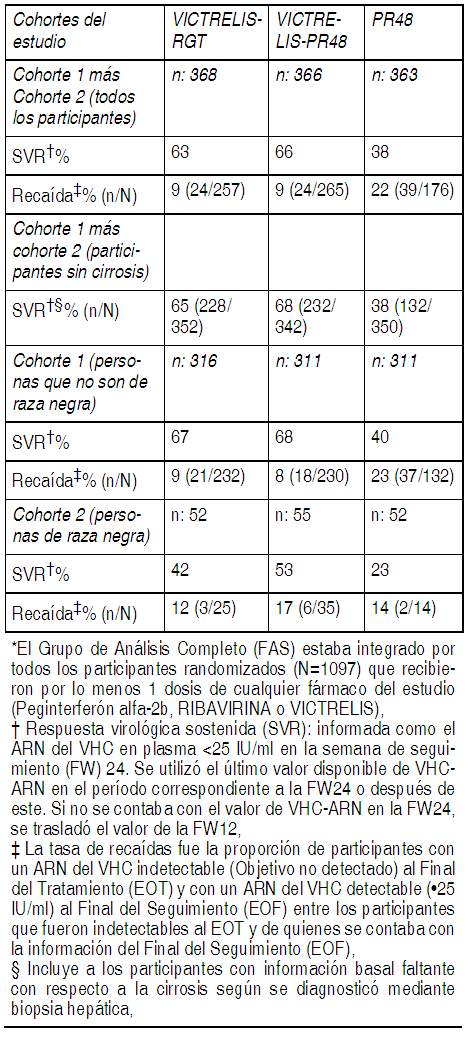{kind=link}
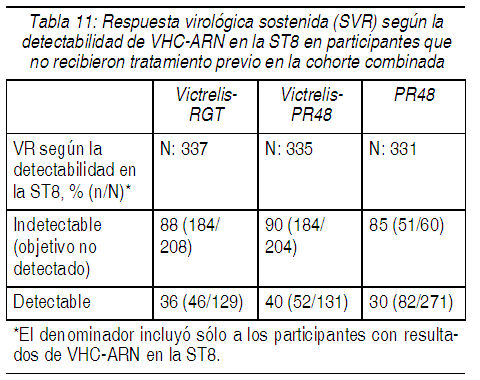{kind=link}
{kind=link}
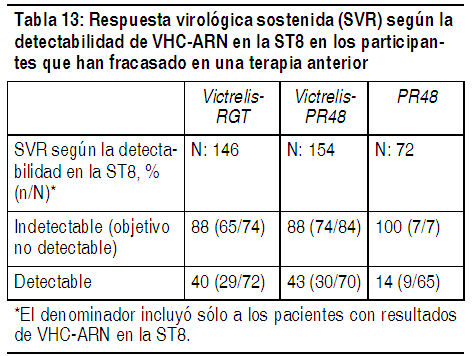{kind=link}
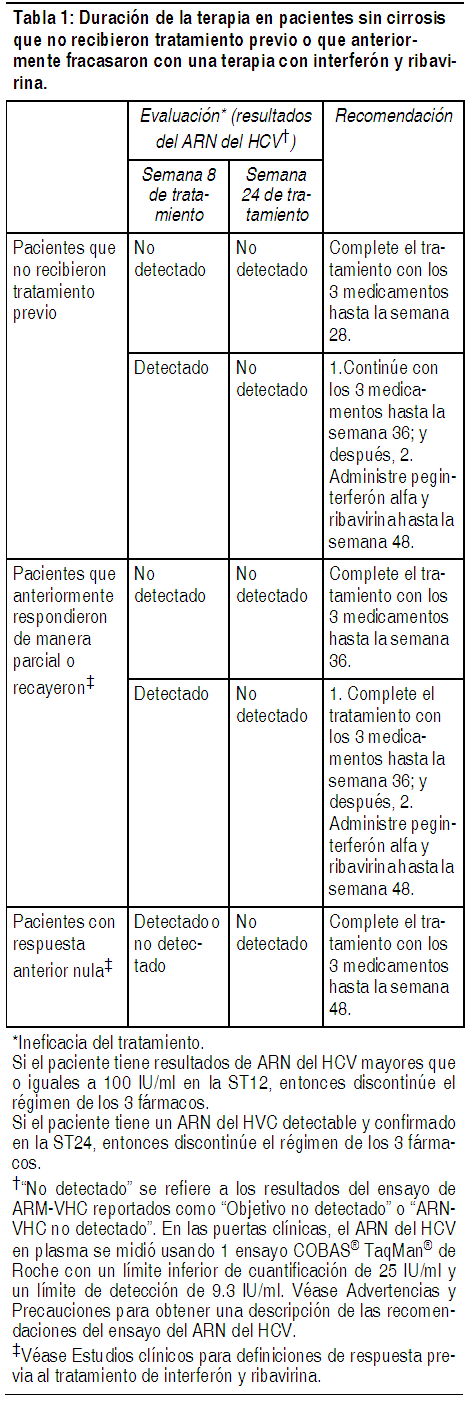{kind=link}
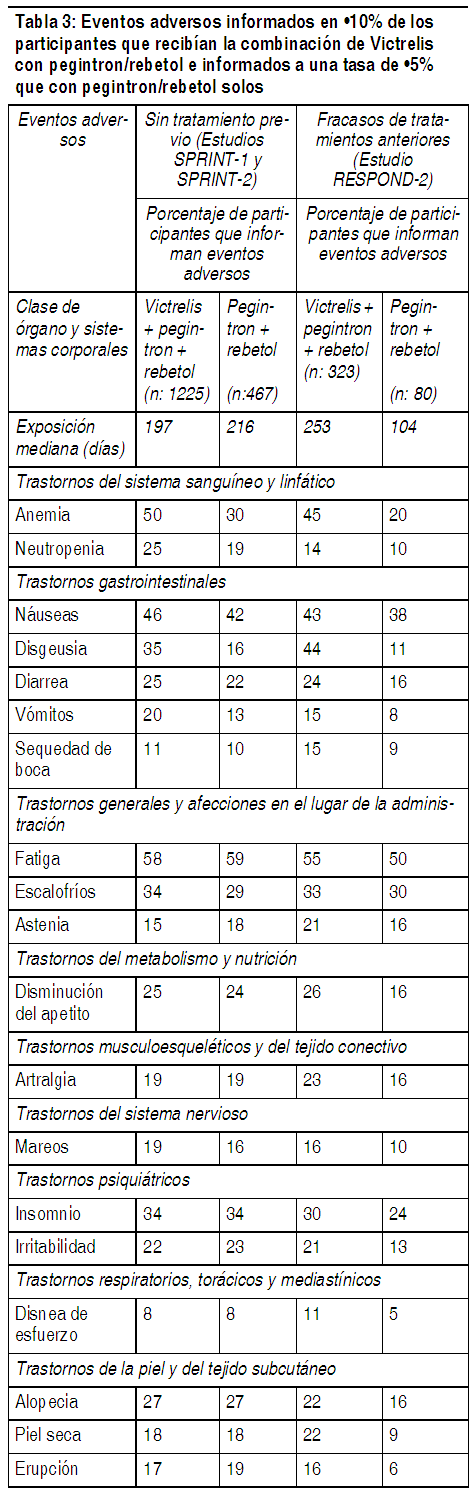{kind=link}
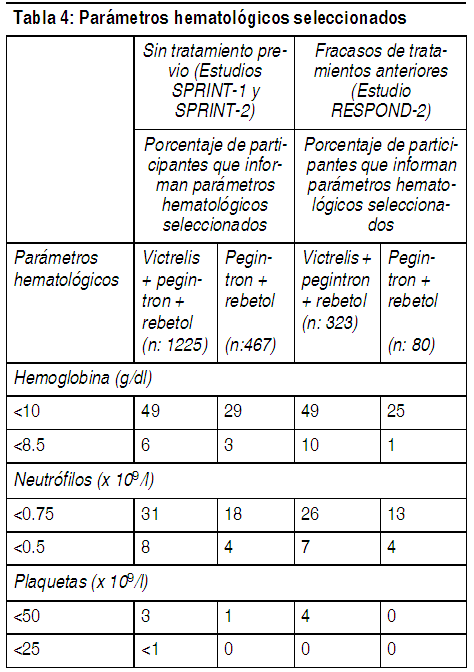{kind=link}
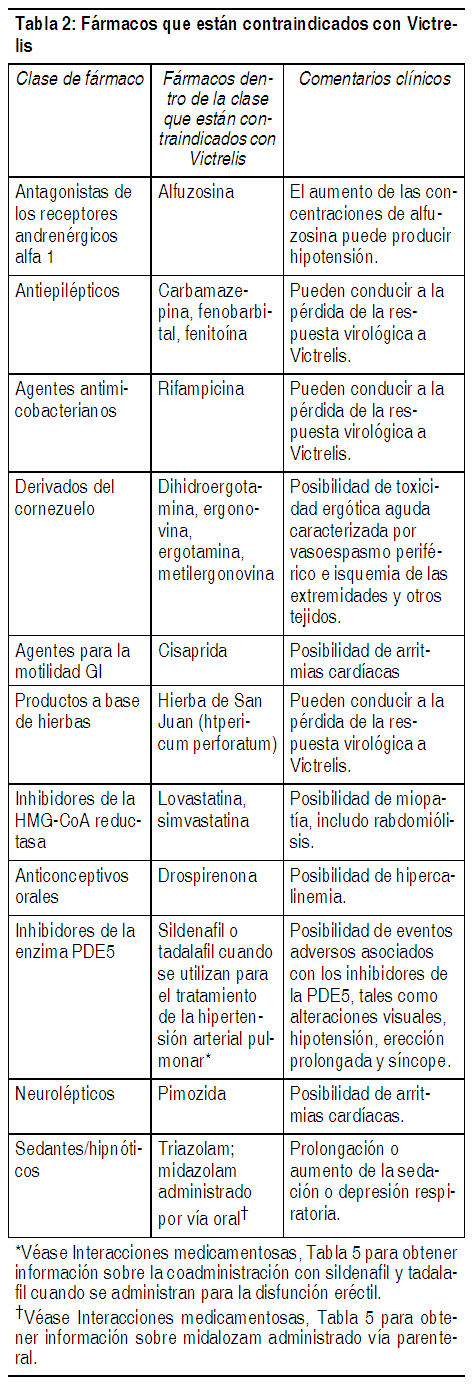{kind=link}