 |
Composición:
Cada comprimido contiene: 40 mg de Regorafenib en el estado Anhidro, lo cual corresponde a 41.49 mg de Regorafenib Monohidrato, y los siguientes ingredientes inactivos: Celulosa Microcristalina, Croscarmelosa de Sodio, Estearato de Magnesio, Povidona, y Dióxido de Silicona Coloidal. El recubrimiento contiene los siguientes ingredientes inactivos: Oxido de Hierro Rojo, Oxido de Hierro Amarillo, Lecitina (Soja), Polietilenglicol 3350, Alcohol Polivinķlico, Talco, y Dióxido de Titanio.
|
|
Descripción:
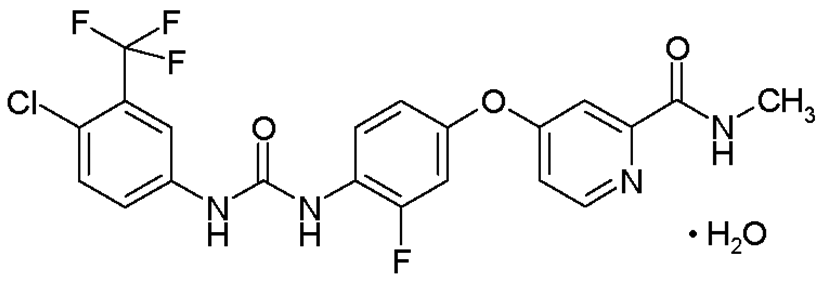
Stivarga es un comprimido recubierto, ovalado, color rosado claro, de 40 mg, marcado con 'BAYER' en una cara y '40' en la otra cara. Stivarga (regorafenib) tiene el nombre quķmico 4-[4-({[4-cloro-3-(trifluorometil)fenil] carbamoil} amino)-3-fluorofenoxi]-N-metilpiridina-2-carboxamida monohidrato. Regorafenib tiene la siguiente fórmula estructural:
Ver Tabla
Regorafenib es un monohidrato y tiene una fórmula molecular C21H15ClF4N4O3 · H2O y un peso molecular de 500.83. Regorafenib es prįcticamente insoluble en agua, ligeramente soluble en acetonitrilo, metanol, etanol, y acetato etķlico y escasamente soluble en acetona. Los comprimidos de Stivarga para administración oral estįn formulados como comprimidos ovalados de color rosado claro marcado con "Bayer" en una cara y "40" en la otra cara.
|
|
Acción Terapéutica:
Antineoplįsico. Código ATC: L01XE21.
|
|
Indicaciones:
Stivarga estį indicado para el tratamiento de pacientes con cįncer colorrectal metastįsico (metastatic colorectal cancer, CRC) que no sean candidatos o hayan sido tratados previamente con quimioterapia basada en fluoropirimidina, o terapias moleculares contra el factor de crecimiento endotelial vascular (Vascular Endothelial Growth Factor, VEGF), y, si presentan el gen KRAS de tipo silvestre o no mutado, con una terapia contra el receptor del factor de crecimiento epidérmico (epidermal growth factor receptor, EGFR). Tratamiento de pacientes con tumores del estroma gastrointestinal (Gastrointestinal Stromal Tumors, GIST) irresecable o metastįsico que fueron tratados previamente con Imatinib mesilato o Sunitinib malato.
|
|
Propiedades:
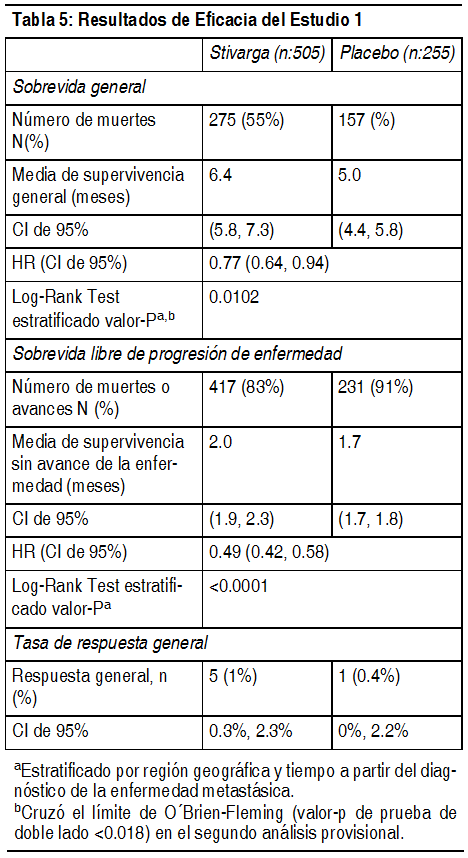
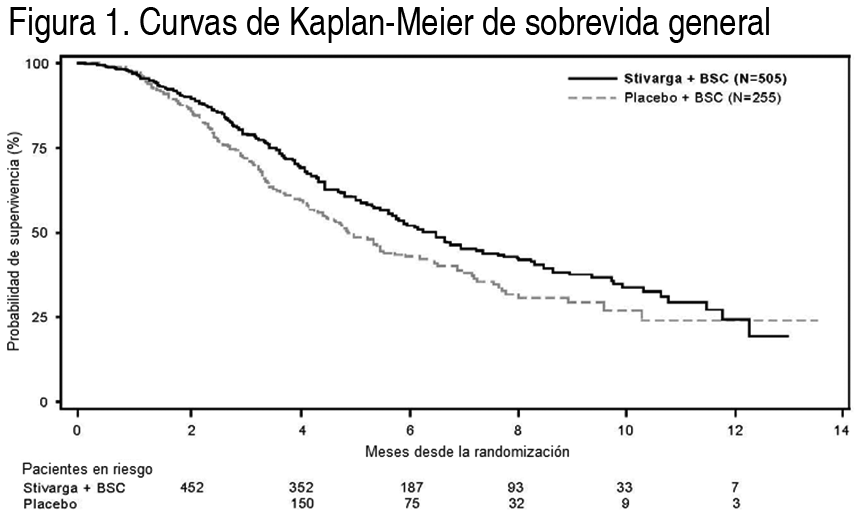
Mecanismo de acción: Regorafenib es un inhibidor molecular de mśltiples quinasas intracelulares y unidas a la membrana celular, involucradas en las funciones celulares normales y en procesos patológicos tales como oncogénesis, angiogénesis tumoral, y mantenimiento del micro ambiente tumoral. En ensayos bioquķmicos o celulares in vitro, regorafenib o sus principales metabolitos activos humanos M-2 y M-5 inhibió la actividad de RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alfa, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAFV600E, SAPK2, PTK5, y Ab1 en concentraciones de regorafenib que han sido logradas clķnicamente. En modelos in vivo, regorafenib demostró actividad anti-angiogénica en un modelo de tumor de rata, una inhibición de crecimiento tumoral asķ como actividad anti-metastįsica en varios modelos de xenoinjerto de ratón incluyendo algunos de carcinoma colorrectal humano. Farmacocinética: Absorción: Después de una dosis śnica de 160 mg de Stivarga en pacientes con tumores sólidos avanzados, regorafenib alcanza un nivel plasmįtico mįximo de la media geométrica (Cmax) de 2.5 µg/ml en una promedio de tiempo de 4 horas y una media geométrica del įrea bajo la curva de concentración plasmįtica frente a tiempo (ABC) de 70.4 g*h/ml. El ABC de regorafenib en estado de equilibrio aumenta menos que proporcionalmente a la dosis con dosis mayores de 60 mg. En estado de equilibrio, regorafenib alcanza una media geométrica de Cmax de 3.9 µg/ml y una media geométrica del ABC de 58.3 µg*h/ml. El coeficiente de variación del ABC y la Cmax es entre 35% y 44%. La biodisponibilidad relativa media de los comprimidos en comparación con una solución oral es 60% a 83%. En 1 estudio de efecto sobre los alimentos, 24 hombres sanos recibieron una dosis śnica de 160 mg de Stivarga en 3 ocasiones separadas: en ayunas, con una comida alta en grasa y con una comida baja en grasa. Una comida alta en grasa (945 calorķas y 54.6 g de grasa) aumentó la media del ABC de regorafenib en 48% y disminuyó la media del ABC de los metabolitos M-2 y M-5 en 20% y 51%, respectivamente, en comparación con el estado de ayuno. Una comida baja en grasa (319 calorķas y 8.2 g de grasa) aumentó la media del ABC de regorafenib, M-2 y M-5 en 36%, 40% y 23%, respectivamente, en comparación con el estado de ayuno. Stivarga se administró con una comida baja en grasa en los Estudios 1 y 2 (véase Posologķa, Estudios clķnicos). Distribución: Regorafenib es sometido a la circulación enterohepįtica con mśltiples picos de concentración plasmįtica observados a en 24 horas del intervalo de la dosis. Regorafenib tiene alta afinidad de unión a las proteķnas de plasma humano (99.5%). Metabolismo: Regorafenib se metaboliza por el CYP3A4 y el UGT1A9. Los principales metabolitos circulantes de regorafenib medidos en estado de equilibrio en plasma humano son M-2 (N-óxido) y M-5 (N-óxido y N-desmetilo), los cuales tienen actividad farmacológica y concentraciones en estado estable in vitro similares a regorafenib. M-2 y M-5 tienen alta afinidad de unión proteķnica (99.8% y 99.95%, respectivamente). Eliminación: Después de una dosis oral śnica de 160 mg de Stivarga, la media geométrica (rango) de vida media de eliminación para regorafenib y el metabolito M-2 en plasma son 28 horas (14 a 58 horas) y 25 horas (14 a 32 horas), respectivamente. M-5 tiene una media (rango) de la vida media de eliminación mįs prolongada de 51 horas (32 a 70 horas). Aproximadamente 71% de la dosis radiomarcada se excretó en las heces (47% como compuesto precursor, 24% como metabolitos) y 19% de la dosis se excretó en la orina (17% como glucurónidos) dentro de 12 dķas después de la administración de una solución oral radiomarcada en una dosis de 120 mg. Pacientes son insuficiencia hepįtica: La farmacocinética de regorafenib, M-2 y M-5 se evaluó en 14 pacientes con carcinoma hepatocelular (HCC) e insuficiencia hepįtica leve (Child-Pugh A); 4 pacientes con HCC e insuficiencia hepįtica moderada (Child-Pugh B); y 10 pacientes con tumores sólidos y función hepįtica normal después de la administración de una dosis śnica de 100 mg de Stivarga. No se observaron diferencias clķnicamente importantes en la media de exposición de regorafenib, M-2, o M-5 en pacientes con insuficiencia hepįtica leve o moderada en comparación con los pacientes con función hepįtica normal. La farmacocinética de regorafenib no ha sido estudiada en pacientes con insuficiencia hepįtica severa (Child-Pugh C). Pacientes con insuficiencia renal: La farmacocinética de regorafenib, M-2 y M-5 se evaluó en 10 pacientes con insuficiencia renal leve (CLcr 60-89 ml/min) y 18 pacientes con función renal normal después de la administración de Stivarga en una dosis diaria de 160 mg durante 21 dķas. No se observaron diferencias en la media de exposición en estado de equilibrio de regorafenib, M-2 o M-5 en pacientes con insuficiencia renal leve en comparación con pacientes con función renal normal. Existen datos limitados de la farmacocinética de pacientes con insuficiencia renal moderada (CLcr 30-59 ml/min). La farmacocinética de regorafenib no ha sido estudiada en pacientes con insuficiencia renal severa o enfermedad renal en etapa terminal. Estudios clķnicos: Cįncer colorrectal: La eficacia y la seguridad clķnica de Stivarga se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doble ciego, controlado con placebo (Estudio 1) en 760 pacientes con cįncer colorrectal metastįsico tratado previamente. La medida de la eficacia de los resultados principales fue la sobrevida general (SG); las medidas de la eficacia de los resultados complementarios incluyeron sobrevida libre de progresión de la enfermedad (SLP) y la tasa de respuesta objetiva del tumor. Los pacientes fueron aleatorizados para recibir una dosis diaria por vķa oral de 160 mg de regorafenib (N=505) mįs el Mejor Cuidado de Soporte (MCS) o placebo (N=255) mįs MCS durante los primeros 21 dķas de cada ciclo de 28 dķas. Stivarga se administró con un desayuno bajo en grasa que contenķa menos de 30% de grasa (véase Posologķa, Farmacologķa clķnica). El tratamiento continuó hasta el avance de la enfermedad o una toxicidad inaceptable. En toda la población aleatorizada la edad promedio fue 61 ańos, 61% fueron hombres, 78% fueron blancos, y todos los pacientes tuvieron un estado funcional del ECOG basal de 0 ó 1. El sitio primario de enfermedad fue el colon (65%), recto (29%), o ambos (6%). Se informó historia de evaluación del KRAS para 729 (96%) pacientes; 430 (59%) de estos pacientes informaron tener mutación del KRAS. La media del nśmero de lķneas de terapia previa para enfermedad metastįsica fue 3. Todos los pacientes recibieron tratamiento previo con terapia en base a fluoropirimidina, oxaliplatino e irinotecįn, y con bevacizumab. Todos excepto uno de los pacientes con tumores negativos para mutación del KRAS recibieron panitumumab o cetuximab. La adición de Stivarga a MCS provocó una aumento estadķsticamente significativo en la supervivencia en comparación con placebo mįs MCS (véase la Tabla 5 y la Figura 1).
Ver Tabla
Ver Tabla
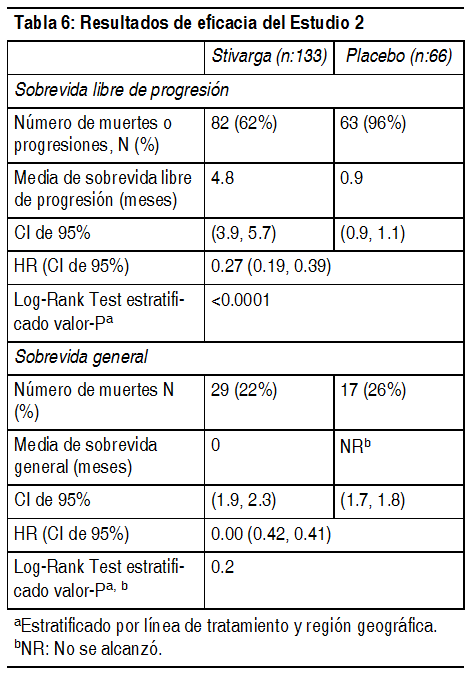
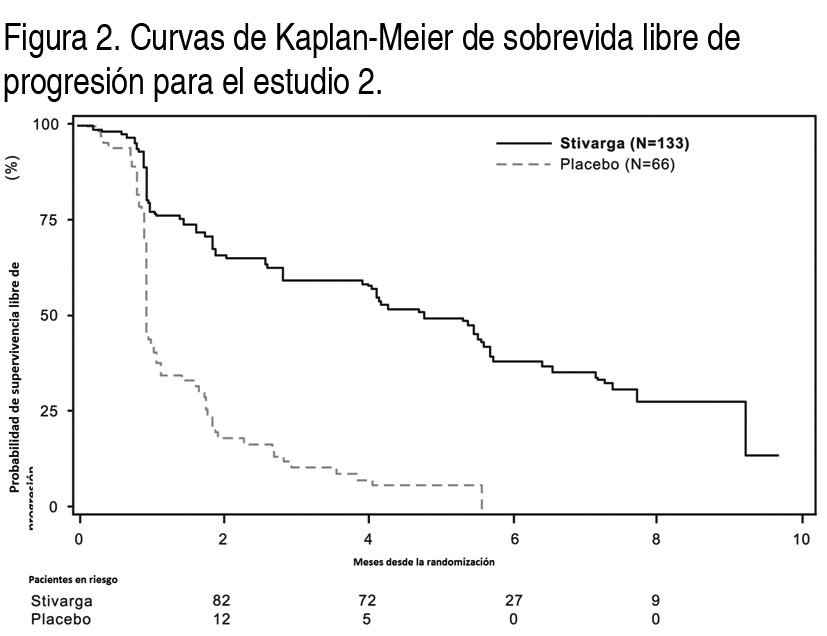
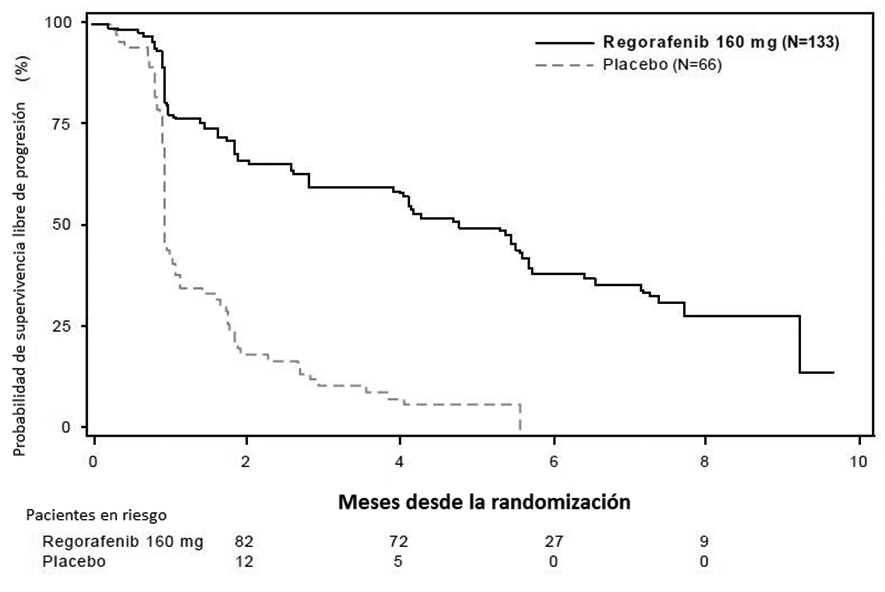
Tumores estromales gastrointestinales (GIST): Se evaluó la eficacia y seguridad de Stivarga en un estudio internacional, multicéntrico, aleatorizado (2:1) doble ciego, controlado con placebo (Estudio 2) en 199 pacientes con tumor estromal gastrointestinal no extirpable, localmente avanzado o metastįsico (GIST) que habķan sido tratados previamente con mesilato de imatinib y malato de sunitinib. Se estratificó la aleatorización por lķnea de terapia (3 vs. 4 o mįs) y región geogrįfica (Asia frente al resto del mundo). La medida de resultado de eficacia mayor del Estudio 2 fue sobrevida libre de progresión (SLP) en base a la evaluación de la enfermedad mediante la revisión radiológica independiente utilizando los criterios modificados de RECIST 1.1, en los cuales los ganglios linfįticos y lesiones óseas no fueron lesiones objetivo y el ganglio tumoral nuevo con crecimiento progresivo dentro de la masa de tumor pre-existente fue la progresión. La medida clave de resultado secundario fue la sobrevida general. Los pacientes fueron aleatorizados para recibir 160 mg de regorafenib oralmente 1 vez al dķa (N=133) mįs el Mejor Cuidado de Soporte (MCS) o placebo (N=66) mįs MCS por los primeros 21 dķas de cada ciclo de 28 dķas. El tratamiento continuó hasta la progresión de la enfermedad o toxicidad inaceptable. En el Estudio 2, la edad promedio de los pacientes fue 60 ańos, 64% fueron hombres, 68% fueron de raza blanca y todos los pacientes tuvieron un estado de rendimiento de ECOG basal de 0 (55%) o 1 (45%). Al momento de la progresión de la enfermedad segśn se evaluó por la revisión central, se rompió el ciego del estudio y todos los pacientes recibieron la oportunidad de tomar Stivarga a criterio del investigador. Cincuenta y seis (85%) pacientes aleatorizados a placebo y 41 (31%) pacientes aleatorizados a Stivarga, recibieron de manera abierta Stivarga. Se demostró una mejora estadķsticamente significativa en SLP entre los pacientes tratados con Stivarga en comparación con el placebo (ver la Tabla 6 y la Figura 2). No hubo ninguna diferencia estadķsticamente significativa en la sobrevida general al momento del anįlisis provisional planeado en base a 29% de los eventos totales para el anįlisis final.
Ver Tabla
Ver Tabla
Ver Tabla
|
|
Posologķa:
Dosis recomendada: La dosis recomendada es 160 mg de regorafenib (4 comprimidos recubiertos de 40 mg) tomado por vķa oral 1 vez al dķa durante los primeros 21 dķas de cada ciclo de 28 dķas. Continuar el tratamiento hasta el avance de la enfermedad o toxicidad inaceptable. Tomar Stivarga a la misma hora cada dķa. Ingerir el comprimido entero con un desayuno bajo en grasa que contenga menos de 30% de grasa (véase Farmacologķa clķnica). Los ejemplos de un desayuno bajo en grasa incluyen 2 rebanadas de pan blanco tostado con 1 cucharada de margarina baja en grasa y 1 cucharada de mermelada, y 8 onzas de leche descremada (319 calorķas y 8.2 g de grasa); o 1 taza de cereal, 8 onzas de leche descremada, 1 rebanada de tostada con mermelada, jugo de manzana, y 1 taza de café o té (520 calorķas y 2 g de grasa). No se debe tomar 2 dosis de Stivarga en el mismo dķa para compensar una dosis perdida del dķa anterior. Modificaciones de la dosis: Interrumpir Stivarga en los siguientes casos: Reacción cutįnea mano-pie (HFSR) [eritrodisestesia palmo-plantar (PPE)] Grado 2 de NCI CTCAE Versión 3.0 (v3.0) que sea recurrente o que no mejora dentro de 7 dķas a pesar de la reducción de la dosis; interrumpir la terapia por un mķnimo de 7 dķas para HFSR Grado 3. Hipertensión sintomįtica Grado 2. Cualquier reacción adversa Grado 3 ó 4 de NCI CTCAE. Reducir la dosis de Stivarga a 120 mg: En el primer caso de HFSR Grado 2 de cualquier duración. Después de la recuperación de cualquier reacción adversa Grado 3 ó 4. En caso de elevación Grado 3 de la aspartato aminotransferasa (AST) y de la alanina aminotransferasa (ALT); reanudar sólo si el beneficio potencial es mayor que el riesgo de hepatotoxicidad. Reducir la dosis de Stivarga a 80 mg: En caso de recurrencia de HFSR Grado 2 con la dosis de 120 mg. Después de la recuperación de cualquier reacción adversa Grado 3 ó 4 con la dosis de 120 mg (excepto hepatotoxicidad). Discontinuar Stivarga permanentemente en los siguientes casos: Fracaso para tolerar la dosis de 80 mg. Cualquier caso de elevación de AST o ALT 20 veces mayor que el lķmite normal superior. Cualquier caso de elevación de AST o ALT 3 veces mayor que el lķmite normal. Superior con elevación concurrente de la bilirrubina 2 veces mayor que el lķmite normal superior. Recurrencia en la elevación de AST o ALT 5 veces mayor que el lķmite normal superior a pesar de la reducción de la dosis a 120 mg. Para cualquier reacción adversa Grado 4; sólo reanudar si el beneficio potencial es mayor que los riesgos.
|
|
Efectos Colaterales:
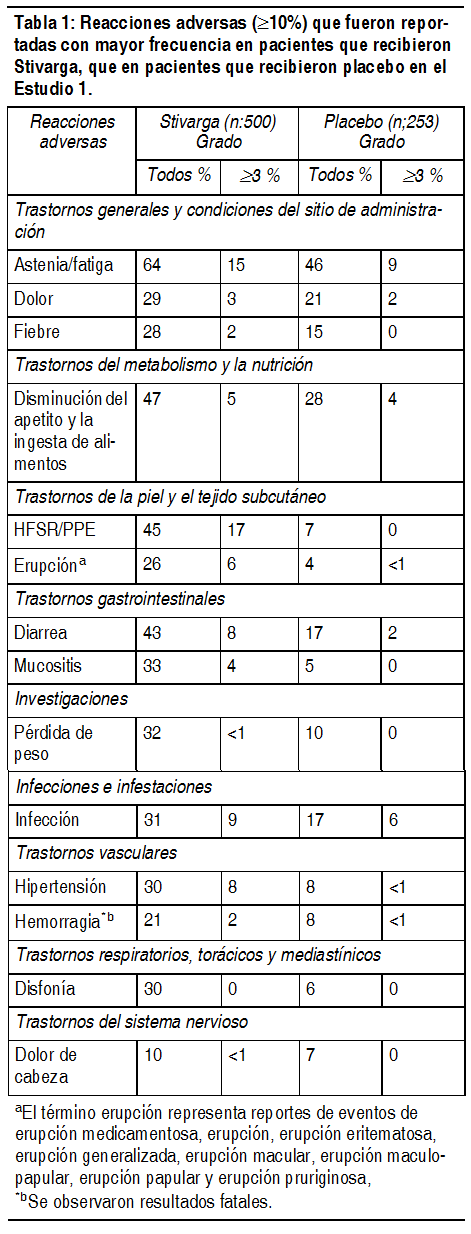
Las siguientes reacciones adversas severas se tratan en otra sección de este inserto: Hepatotoxicidad (véase Advertencias y Precauciones). Hemorragia (véase Advertencias y Precauciones). Toxicidad dermatológica (véase Advertencias y Precauciones). Hipertensión (véase Advertencias y Precauciones). Isquemia e infarto de miocardio (véase Advertencias y Precauciones). Sķndrome de leucoencefalopatķa posterior reversible (RPLS) (véase Advertencias y Precauciones). Perforación o fķstula gastrointestinal (véase Advertencias y Precauciones). Debido a que los ensayos clķnicos se realizan bajo condiciones ampliamente variables, las tasas de reacciones adversas observados en los ensayos clķnicos de un fįrmaco no se pueden comparar directamente con las tasas de reacciones adversas de los ensayos clķnicos de otro fįrmaco y pueden no reflejar las tasas observadas en la prįctica. Las reacciones farmacológicas adversas observadas mįs frecuentemente ( ³ 20%) en los pacientes que reciben Stivarga son astenia/fatiga, HFSR, diarrea disminución del apetito, disminución del consumo de alimentos, hipertensión, mucositis, disfonķa, infección, dolor (no especificado de otro modo), pérdida de peso, dolor gastrointestinal y abdominal, erupción, fiebre y nįuseas. Las reacciones farmacológicas adversas mįs severas en pacientes que recibe Stivarga fueron hepatotoxicidad, hemorragia y perforación gastrointestinal. Experiencia en los ensayos clķnicos: Cįncer colorrectal: Los datos de seguridad descritos a continuación a excepción que se indiquen en una nota aclaratoria, derivan de un ensayo aleatorizado doble ciego controlado con placebo, (Estudio 1) en el cual 500 pacientes (edad media 61 ańos; 61% hombres) con cįncer colorrectal metastįsico tratado previamente recibieron Stivarga como un agente śnico con la dosis diaria de 160 mg durante las primeras 3 semanas de cada ciclo de tratamiento de 4 semanas y 253 pacientes (edad media 61 ańos; 60% hombres) recibieron placebo. La duración promedio de la terapia fue 7.3 semanas (rango 0.3, 47.0) para los pacientes que recibieron Stivarga. Debido a las reacciones adversas, 61% de los pacientes que recibieron Stivarga requirieron una interrupción de la dosis y a 38% de los pacientes se les redujo la dosis. Las reacciones adversas relacionadas con el fįrmaco que resultaron en descontinuación del tratamiento fueron reportados en 8.2% de pacientes tratados con Stivarga en comparación con 1.2% de los pacientes que recibieron placebo. La reacción cutįnea mano-pie (HFSR) y la erupción fueron las razones mįs comunes para la descontinuación permanente de Stivarga. La Tabla 1 compara la incidencia de las reacciones adversas ( ³ 10%) que fueron reportadas con mayor frecuencia en pacientes que recibieron Stivarga, que en pacientes que recibieron placebo (Estudio 1).
Ver Tabla
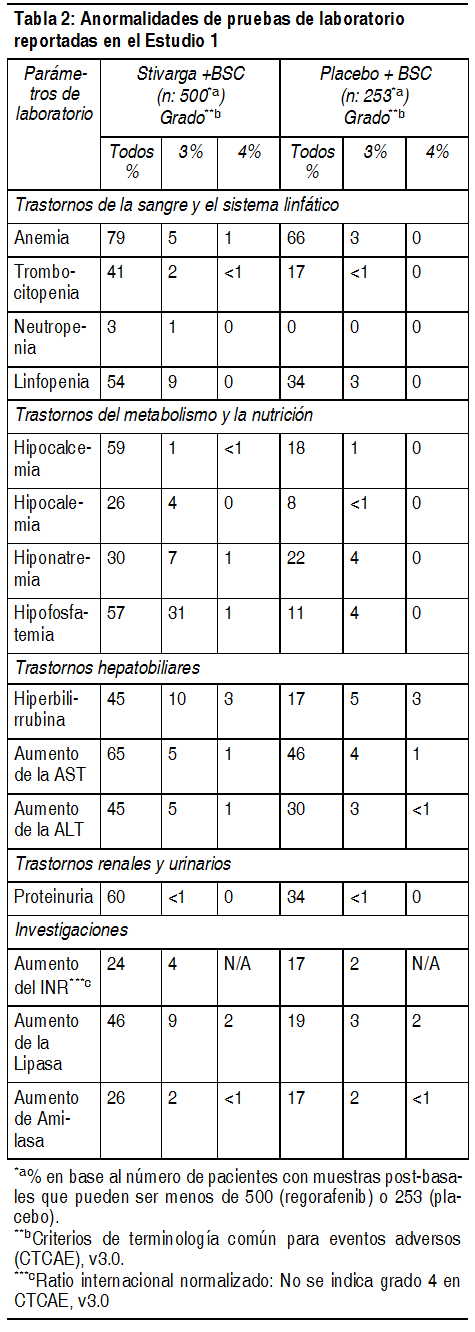
Anormalidades de laboratorio: Las anormalidades de laboratorio observadas en el Estudio 1 se muestran en la Tabla 2.
Ver Tabla
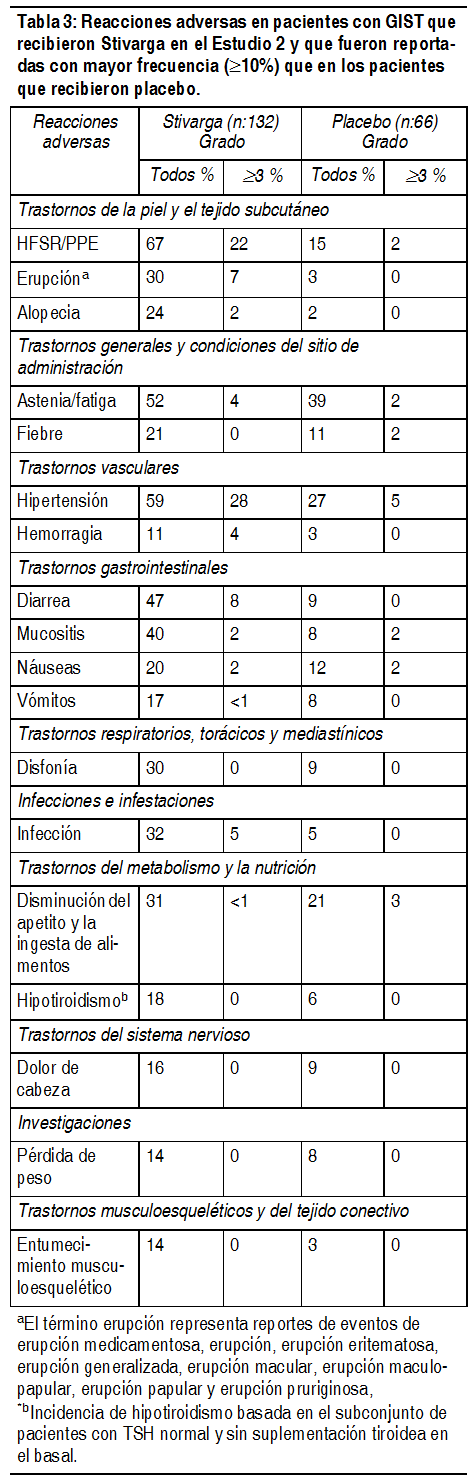
Tumores estromales gastrointestinales (GIST): Los datos de seguridad descritos a continuación son derivados de un ensayo aleatorizado doble ciego controlado con placebo, (2:1) (Estudio 2) en la cual 132 pacientes (edad media 60 ańos, 64% hombres) con GIST tratados previamente recibieron Stivarga como un agente śnico a una dosis de 160 mg al dķa por las primeras 3 semanas de cada ciclo de 4 semanas de tratamiento y 66 pacientes (edad media 61 ańos, 64% hombres) recibieron placebo. El promedio de duración de la terapia fue 22.9 semanas (rango 0.1, 50.9) en los pacientes que recibieron Stivarga. Se requirió interrupciones de la dosis durante los eventos adversos en 58% de los pacientes que recibieron Stivarga y en 50% de los pacientes se les redujo la dosis. Las reacciones adversas relacionadas al fįrmaco que resultaron en descontinuación del tratamiento fueron reportadas en 2.3% de los pacientes tratados con Stivarga en comparación con 1.5% de los pacientes que recibieron placebo. La Tabla 3 compara la incidencia de reacciones adversas en pacientes con GIST que recibieron Stivarga y que fueron reportadas con mayor frecuencia ( ³ 10%) que en los pacientes que recibieron placebo (Estudio 2).
Ver Tabla
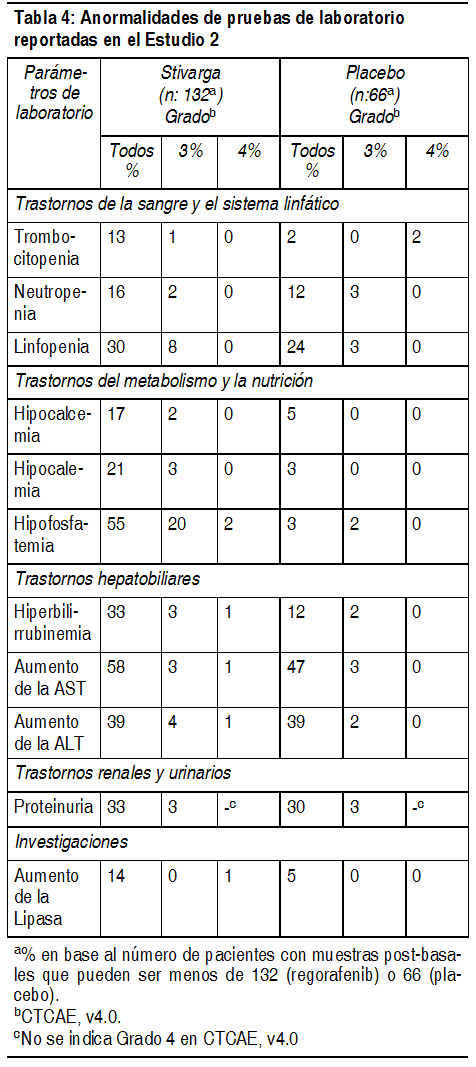
Anormalidades de laboratorio: Las anormalidades de laboratorio observadas en el Estudio 2 se muestran en la Tabla 4.
Ver Tabla
|
|
Contraindicaciones:
Ninguna.
|
|
Advertencias:
Hepatotoxicidad: Se ha observado hepatotoxicidad severa y a veces fatal en ensayos clķnicos. Monitorizar la función hepįtica antes y durante el tratamiento. Interrumpir y luego reducir o descontinuar Stivarga si se manifiesta hepatotoxicidad mediante pruebas de función hepįtica elevada o necrosis hepatocelular, dependiendo de la severidad y persistencia. Hepatotoxicidad: Injuria hepįtica severa inducida por el fįrmaco ocurrió con resultado fatal en 0.3% de 1200 pacientes tratados con Stivarga en todos los ensayos clķnicos. Los resultados de biopsia de hķgado, cuando estuvieron disponibles, mostraron necrosis de hepatocitos con infiltración de linfocitos. En el Estudio 1, ocurrió insuficiencia hepįtica fatal en 1.6% de pacientes en el brazo de regorafenib y en 0.4% de pacientes en el brazo de placebo; todos los pacientes con insuficiencia hepįtica tuvieron enfermedad metastįsica en el hķgado. En el Estudio 2, se produjo insuficiencia hepįtica fatal en 0.8% de pacientes en el brazo de regorafenib (ver Reacciones adversas). Obtener pruebas de función hepįtica (ALT, AST y bilirrubina) antes del inicio de la administración Stivarga y monitorear al menos cada 2 semanas durante los primeros 2 meses de tratamiento. De ahķ en adelante, monitorear mensualmente o con mayor frecuencia segśn sea indicado clķnicamente. Monitorear las pruebas de función hepįtica semanalmente en pacientes que han experimentado pruebas de función hepįtica elevada hasta que mejore a menos de 3 veces el lķmite normal superior o el basal. Suspender temporalmente y luego reducir o discontinuar de modo permanente Stivarga dependiendo de la severidad y persistencia de la hepatotoxicidad segśn se manifieste mediante pruebas de función hepįtica elevada o necrosis hepatocelular (véase Posologķa). Hemorragia: Stivarga provocó un aumento en la incidencia de hemorragia. La incidencia general (Grados 1-5) fue de 21% y 11% en pacientes tratados con Stivarga en comparación con 8% y 3% en pacientes tratados con placebo en los Estudios 1 y 2. Ocurrió hemorragia fatal en 4 de 632 (0.6%) pacientes tratados con Stivarga en los Estudios 1 y 2 e implicaron las vķas respiratorias, gastrointestinales o genitourinarias. Discontinuar permanentemente Stivarga en pacientes con hemorragia severa o que amenaza la vida. Monitorear los niveles de INR con mayor frecuencia en pacientes que reciben warfarina (véase Farmacologķa clķnica). Toxicidad dermatológica: Stivarga provocó un aumento en la incidencia de reacciones adversas en piel y tejido subcutįneo (72% frente a 24% en el Estudio 1 y 78% frente a 24% en el Estudio 2), incluyendo reacción cutįnea mano-pie (HFSR) también conocida como eritro-disestesia palmo-plantar (PPE) y erupción severa que requerirį modificación de la dosis. La incidencia general de HFSR fue mayor en los pacientes tratados con Stivarga (45% frente a 7% en el Estudio 1 y 67% frente a 12% en el Estudio 2) que en los pacientes tratados con placebo. La mayorķa de los casos de HFSR en pacientes tratados con Stivarga apareció durante el primer ciclo de tratamiento (69% y 71% de los pacientes que desarrolló HFSR en el Estudio 1 y Estudio 2, respectivamente). La incidencia de HFSR de Grado 3 (17% frente a 0%) en el Estudio 1 y 22% frente a 0% en el Estudio 2, erupción cutįnea grado 3 (6% frente < 1% en el Estudio 1 y 7% frente a 0% en el Estudio 2), reacciones adversas serias de eritema multiforme (0.2% vs. 0% en el Estudio 1) y sķndrome de Stevens Johnson (0.2% frente a 0% en el Estudio 1) fue mįs alta en los pacientes tratados con Stivarga (véase Efectos colaterales). Se produjo necrólisis epidérmica tóxica en 0.17% de 1200 pacientes tratados con Stirvarga entre todos los ensayos clķnicos. La suspensión de Stivarga, la reducción de la dosis o su descontinuación permanentemente, depende de la severidad y persistencia de la toxicidad dermatológica (véase Posologķa). Se debe instituir medidas de apoyo para el alivio sintomįtico. Hipertensión: Stivarga provocó un aumento en la incidencia de hipertensión (30% frente a 8% en el Estudio 1 y 59% frente a 27% en el Estudio 2) (véase Efectos colaterales). Ocurrió crisis hipertensiva en 0.25% de 1200 pacientes tratados con Stivarga a través de todos los ensayos clķnicos. El inicio de la hipertensión ocurrió en el primer ciclo de tratamiento en la mayorķa de pacientes que desarrollaron hipertensión (72% en el Estudio 1 y Estudio 2). No iniciar Stivarga a menos que la presión arterial esté controlada adecuadamente. Monitorear la presión arterial semanalmente durante las primeras 6 semanas de tratamiento y luego cada ciclo, o con mayor frecuencia, segśn sea indicado clķnicamente. Suspender Stivarga de modo temporal o permanente debido a hipertensión severa o no controlada (véase Posologķa). Isquemia e infarto de miocardio: Stivarga aumentó la incidencia de isquemia e infarto de miocardio en el Estudio 1 (1.2% frente a 0.4%) (véase Efectos colaterales). Suspender en pacientes que desarrollan isquemia o infarto agudo de miocardio. Reanudar Stivarga sólo después de la terminación de eventos isquémicos cardķacos agudos si los beneficios potenciales son mayores que los riesgos de nueva isquemia de miocardio. Sķndrome de leucoencefalopatķa posterior reversible (RPLS): El sķndrome de leucoencefalopatķa posterior reversible (RPLS) un sķndrome de un edema vasogénico subcortical diagnosticado por hallazgos caracterķsticos en la RMN, se produjeron en uno de 1200 pacientes tratados con Stivarga a través de todos los ensayos clķnicos. Realizar una evaluación para RPLS en cualquier paciente que presente convulsiones, cefalea, alteraciones visuales, confusión o alteración de la función mental. Discontinuar Stivarga en pacientes que desarrollan RPLS. Perforación o fķstula gastrointestinal: Ocurrió perforación o fķstula gastrointestinal en 0.6% de 1200 tratados con Stivarga a través de todos los ensayos clķnicos, esto incluyó 4 eventos fatales. En el estudio 2, 2.1% (4/188) de pacientes tratados con Stivarga que fueron tratados durante la parte ciega o abierta del estudio desarrollaron fķstula o perforación gastrointestinal, de éstos, dos casos de perforación gastrointestinal fueron fatales. Discontinuar Stivarga permanentemente en pacientes que desarrollan perforación o fķstula gastrointestinal. Complicaciones en la cicatrización de heridas: No se han realizado estudios formales del efecto de regorafenib sobre la cicatrización de las heridas. Debido a que los inhibidores del receptor del factor de crecimiento endotelial vascular (VEGFR) como regorafenib podrķa impedir la cicatrización de heridas, el tratamiento con regorafenib debe interrumpirse al menos 2 semanas antes de una cirugķa programada. La decisión para reanudar regorafenib después de cirugķa debe hacerse en base al criterio clķnico de cicatrización adecuada de la herida. Se debe discontinuar regorafenib en pacientes con dehiscencia de la herida. Toxicidad embriofetal: Stivarga puede provocar dańo fetal cuando se administra a una mujer embarazada. Regorafenib fue letal y teratogénico a los embriones de ratas y conejos que fueron expuestos a dosis menores que la dosis recomendada a humanos, con aumento de incidencias de malformaciones cardiovasculares, genitourinarias y esqueléticas. Si este fįrmaco se usa durante el embarazo, o si la paciente queda embarazada mientras que toma este fįrmaco, la paciente debe considerar el riesgo potencial para el feto (véase Uso en poblaciones especķficas).
|
|
Precauciones:
Uso en poblaciones especķficas: Embarazo: Embarazo Categorķa D (véase Advertencias y Precauciones). Resumen de riesgos: Basado en su mecanismo de acción, Stivarga puede provocar dańo fetal cuando se administra a una mujer embarazada. No existen estudios adecuados y bien controlados con Stivarga en mujeres embarazadas. Regorafenib fue teratogénico y letal a los embriones de ratas y conejos que fueron expuestos a dosis menores de las de las dosis recomendadas en humanos, con aumento en las incidencias de malformaciones cardiovasculares, genitourinarias y esqueléticas. Si este fįrmaco se usa durante el embarazo o si la paciente queda embarazada mientras toma este fįrmaco, se debe advertir a la paciente sobre el riesgo potencial para el feto. Datos en animales: En estudios de desarrollo embriofetal, se observó un pérdida total del embarazo (100% de resorción de la crķa) en ratas con dosis tan bajas como 1 mg/kg (aproximadamente 6% de la dosis recomendada en humanos, en base al įrea de superficie corporal) y en conejos con dosis tan bajas como 1.6 mg/kg (aproximadamente 25% de la exposición humana a la dosis clķnicamente recomendada medida mediante el ABC Įrea bajo la curva). En 1 estudio de distribución de dosis śnica en ratas gestantes, hubo un aumento de la penetración de regorafenib a través de la barrera hematoencefįlica en fetos en comparación con las madres. En un estudio de dosis repetidas con administración diaria de regorafenib a ratas gestantes durante la organogénesis, los hallazgos incluyeron osificación retardada en fetos con dosis ³ 0.8 mg/kg (aproximadamente 5% de la dosis recomendada en humanos, en base al įrea de superficie corporal) con aumentos dosis dependientes en malformaciones esqueléticas incluyendo paladar hendido y fontanela agrandada a dosis ³ 1 mg/kg (aproximadamente 10% de la exposición clķnica en base al ABC). A dosis ³ 1.6 mg/kg (aproximadamente 11% de la dosis recomendada en humanos en base al įrea de superficie corporal), hubieron aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares, anomalķas externas, hernia diafragmįtica, y dilatación de la pelvis renal. En conejas preńadas a las que se les administró regorafenib diariamente durante la organogénesis, hubo hallazgos de defectos del septum ventricular evidentes en la menor dosis sometida a prueba de 0.4 mg/kg (aproximadamente 7% del ABC en pacientes con la dosis recomendada). En dosis ³ 0.8 mg/kg (aproximadamente 15% de la exposición humana con la dosis recomendada en humanos en base al ABC), la administración de regorafenib provocó aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares adicionales y anomalķas esqueléticas asķ como efectos adversos significativos en el sistema urinario incluyendo agenesia renal/uréter; rińón pequeńo, deformado y mal posicionado; e hidronefrosis. La proporción de fetos viables que fueron machos disminuyó con el aumento de la dosis en dos estudios de toxicidad embriofetal en conejos. Madres en perķodo de lactancia: Se desconoce si regorafenib o sus metabolitos se excretan en la leche materna humana. En ratas, regorafenib y sus metabolitos se excretan en la leche. Debido a que muchos fįrmacos se excretan en la leche materna y debido al potencial para reacciones adversas severas de Stivarga en lactantes, se debe tomar una decisión con respecto a descontinuar la lactancia o descontinuar el fįrmaco, tomando en cuenta la importancia del fįrmaco para la madre. Uso pediįtrico: La seguridad y la eficacia de Stivarga en pacientes pediįtricos menores de 18 ańos de edad no han sido establecidas. En estudios de dosis repetidas de 28 dķas en ratas se observaron hallazgos dependientes de la dosis de alteración de la dentina y estasis vascular. Estos hallazgos se observaron en dosis de regorafenib tan bajas como 4 mg/kg (aproximadamente 25% del ABC en humanos con la dosis recomendada). En estudios de dosis repetidas de 13 semanas en perros se observaron hallazgos similares de alteración dentina en dosis tan bajas como 20 mg/kg (aproximadamente 43% del ABC en humanos con la dosis recomendada). La administración de regorafenib en estos animales también provocó un crecimiento y engrosamiento persistente de la placa de crecimiento de la epķfisis femoral. Uso geriįtrico: De los 632 pacientes tratados con Stivarga quienes fueron enrolados en los Estudios 1 y 2, el 37% tenķan 65 ańos de edad y mįs, mientras que 8% tenķan 75 ańos de edad y mįs. No se observaron diferencias generales en la seguridad y eficacia entre estos pacientes y los pacientes mįs jóvenes. Insuficiencia hepįtica: No se observaron diferencias clķnicamente importantes en la media de exposición de regorafenib o los metabolitos activos M-2 y M-5 en pacientes con carcinoma hepatocelular e insuficiencia hepįtica leve (Child-Pugh A) o moderada (Child-Pugh B) en comparación con pacientes con función hepįtica normal (véase Farmacologķa clķnica). No se recomienda ningśn ajuste de dosis en pacientes con insuficiencia hepįtica leve o moderada. Monitorear cuidadosamente a los pacientes con insuficiencia hepįtica con respecto a reacciones adversas (véase Advertencias y Precauciones). El uso de Stivarga no se recomienda en pacientes con insuficiencia hepįtica severa (Child-Pugh Clase C) debido a que no ha estudiado en esta población. Insuficiencia renal: No se observaron diferencias clķnicamente relevantes en la media de exposición de regorafenib y los metabolitos activos M-2 y M-5 en pacientes con insuficiencia renal leve (CLcr 60-89 ml/min) en comparación con pacientes con función renal normal después de la administración diaria de 160 mg de regorafenib durante 21 dķas (véase Farmacologķa clķnica). No se recomienda ningśn ajuste de dosis en pacientes con insuficiencia renal leve. Los datos farmacocinéticos son limitados hasta pacientes con insuficiencia renal moderada (CLcr 30-59 ml/min). No existen estudios de Stivarga en pacientes con insuficiencia renal severa o enfermedad renal en estadķo terminal. Mujeres y hombres con potencial reproductivo: Anticoncepción: Utilice un método anticonceptivo eficaz durante el tratamiento y hasta 2 meses después de la finalización del tratamiento. Infertilidad: No existen datos sobre el efecto de Stivarga sobre la fertilidad humana. Los resultados de estudios en animales indican que regorafenib puede afectar la fertilidad masculina y femenina (véase Toxicologķa no clķnica). Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios que examinan el potencial carcinogénico de regorafenib. Regorafenib por sķ mismo no demostró genotoxicidad en ensayos in vitro o in vivo; sin embargo, un metabolito activo humano principal de regorafenib, (M-2), fue positivo para clastogenicidad, provocando aberración cromosómica en células de hįmster chino V79. No se han realizado estudios dedicados a examinar los efectos de regorafenib sobre la fertilidad; sin embargo, hubo hallazgos histológicos de atrofia tubular y degeneración de los testķculos, atrofia en la vesķcula seminal, y residuos celulares y oligoespermia en los epidķdimos en ratas machos en dosis similares a aquellas en humanos en las dosis clķnicas recomendadas en base al ABC. En ratas hembras, hubo aumento en los hallazgos de necrosis de cuerpo lśteo en los ovarios en las mismas exposiciones. Hubo hallazgos similares en perros de ambos sexos en estudios de dosis repetidas en exposiciones de aproximadamente 83% de la exposición humana con la dosis recomendada en humanos en base al ABC. Estos hallazgos sugieren que regorafenib pueden afectar adversamente la fertilidad en humanos. Toxicologķa y/o farmacologķa animal: En un estudio de dosis crónica repetida de 26 semanas en ratas, hubo un aumento dependiente de la dosis en el hallazgo de engrosamiento de la vįlvula aurķculo-ventricular. En una dosis que resultó en una exposición de aproximadamente 12% de la exposición humana con la dosis recomendada, este hallazgo estuvo presente en la mitad de los animales examinados.
|
|
Interacciones Medicamentosas:
Efecto de potentes inductores de CYP3A4 sobre regorafenib: La coadministración de un potente inductor de CYP3A4 (rifampicina) con una dosis śnica de 160 mg de Stivarga disminuyó la media de exposición de regorafenib, aumentó la media de exposición del metabolito activo M-5, y no provocó ningśn cambio en la media de exposición del metabolito activo M-2. Se debe evitar el uso concomitante de Potentes Inductores de CYP3A4 (por ejemplo, rifampicina, fenitoķna, carbamazepina, fenobarbital, y Hierba de San Juan (véase Farmacologķa clķnica). Efecto de potentes inhibidores de CYP3A4 sobre regorafenib: La coadministración de un potente inhibidor de CYP3A4 (ketoconazol) con una dosis śnica de 160 mg de Stivarga aumentó la media de exposición de regorafenib, y disminuyó la media de exposición de los metabolitos activos M-2 y M-5. Se debe evitar el uso concomitante de potentes inhibidores de actividad del CYP3A4 (por ejemplo, claritromicina, jugo de toronja, itraconazol, ketoconazol, posaconazol, telitromicina, y voriconazol) (véase Farmacologķa clķnica). Interacciones medicamentosas: Detección in vitro en enzimas del citocromo P450: Estudios in vitro con microsomas hepįticos humanos o enzimas recombinantes demostraron que regorafenib inhibe competitivamente CYP2C8, CYP2C9, CYP2B6, CYP3A4, y CYP2C19 con valores R1 > 1.1; M-2 inhibe CYP2C9, CYP2C8, CYP3A4, y CYP2D6 con valores R1 > 1.1 y M-5 inhibe CYP2C8 con un valor R1 > 1.1. Estudios in vitro con hepatocitos primarios humanos demostraron que no se espera que regorafenib induzca la actividad enzimįtica de CYP1A2, CYP2B6, CYP2C19, y CYP3A4. Detección in vitro en uridina difosfato glucuronosiltransferasas: Estudios in vitro con microsomas hepįticos humanos demostraron que regorafenib, M-2 y M-5 inhibe competitivamente UGT1A9 y UGT1A1 en concentraciones terapéuticamente relevantes. Detección in vitro en transportadores: Datos in vitro demostraron que regorafenib es un inhibidor de ABCG2 (Proteķna de Resistencia de Cįncer de Mama) y ABCB1 (Glicoproteina-P). Efecto de potentes inductores de CYP3A4 sobre regorafenib: Veintidós hombres sanos recibieron una dosis śnica de 160 mg de Stivarga solo y luego 7 dķas después de iniciar rifampicina. La rifampicina es un potente inductor de CYP3A4, se administró en una dosis diaria de 600 mg durante 9 dķas. La media del ABC de regorafenib disminuyó en 50% y la media del ABC de M-5 aumentó en 264%. No se observó ningśn cambio en la media del ABC de M-2. Efecto de potentes inhibidores de CYP3A4 sobre regorafenib: Dieciocho hombres sanos recibieron una dosis śnica de 160 mg de Stivarga solo y luego 5 dķas después de iniciar ketoconazol. Ketoconazol, un potente inhibidor de CYP3A4, se administró en una dosis diaria de 400 mg durante 18 dķas. La media del ABC de regorafenib aumentó en 33% y la media del ABC de M-2 y M-5 disminuyó en ambos casos en 93%. Efecto de regorafenib sobre un sustrato de sustratos de UGT1A1: Once pacientes recibieron quimioterapia de combinación que contenķa irinotecįn con Stivarga en una dosis de 160 mg. La media del ABC de irinotecįn aumentó en 28% y la media del ABC de SN-38 aumentó en 44% cuando se administró irinotecįn 5 dķas después de la śltima de las 7 dosis diarias de Stivarga.
|
|
Sobredosificación:
La mayor dosis de Stivarga estudiada clķnicamente es 220 mg por dķa. En caso de sospecha de sobredosis, interrumpir Stivarga, instituir tratamiento de soporte y observar hasta la estabilización clķnica.
|
|
Conservación:
Almacenar los comprimidos en el frasco original y no retirar el desecante. Mantener el frasco cerrado herméticamente después de abrir por primera vez. No se exponga a temperaturas superiores a 30ŗC. Desechar cualquier comprimido que no se usó 28 dķas después de abrir el frasco. Eliminar los comprimidos que no se usaron de acuerdo con los requisitos locales. Manténgase fuera del alcance de los nińos. No utilizar después de la fecha de caducidad impresa en el empaque.
|
|
Presentaciones:
Los comprimidos de Stivarga se suministran en empaques conteniendo 3 frascos; cada frasco contiene 28 comprimidos (84 comprimidos por empaque).
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}