 |
Composición:
Cada vial contiene: 120 mg de Denosumab en una solución de 1.7 ml (70 mg/ml). Forma farmacéutica: Solución para inyección subcutánea. Solución transparente, incolora a ligeramente amarilla, y que puede contener oligoelementos proteicos translúcidos a blancos. Lista de excipientes: Sorbitol. Acido Acético Glacial*. Hidróxido de Sodio. Agua para Inyecciones. * Buffer de Acetato está formado por la mezcla de Acido Acético con Hidróxido de Sodio.
|
|
Indicaciones:
Xgeva está indicado en la prevención de eventos esqueléticos relacionados en pacientes con metástasis óseas de tumores sólidos. Limitación de uso importante: Xgeva no está indicado para la prevención de eventos esqueléticos en pacientes con mieloma múltiple.
|
|
Propiedades:
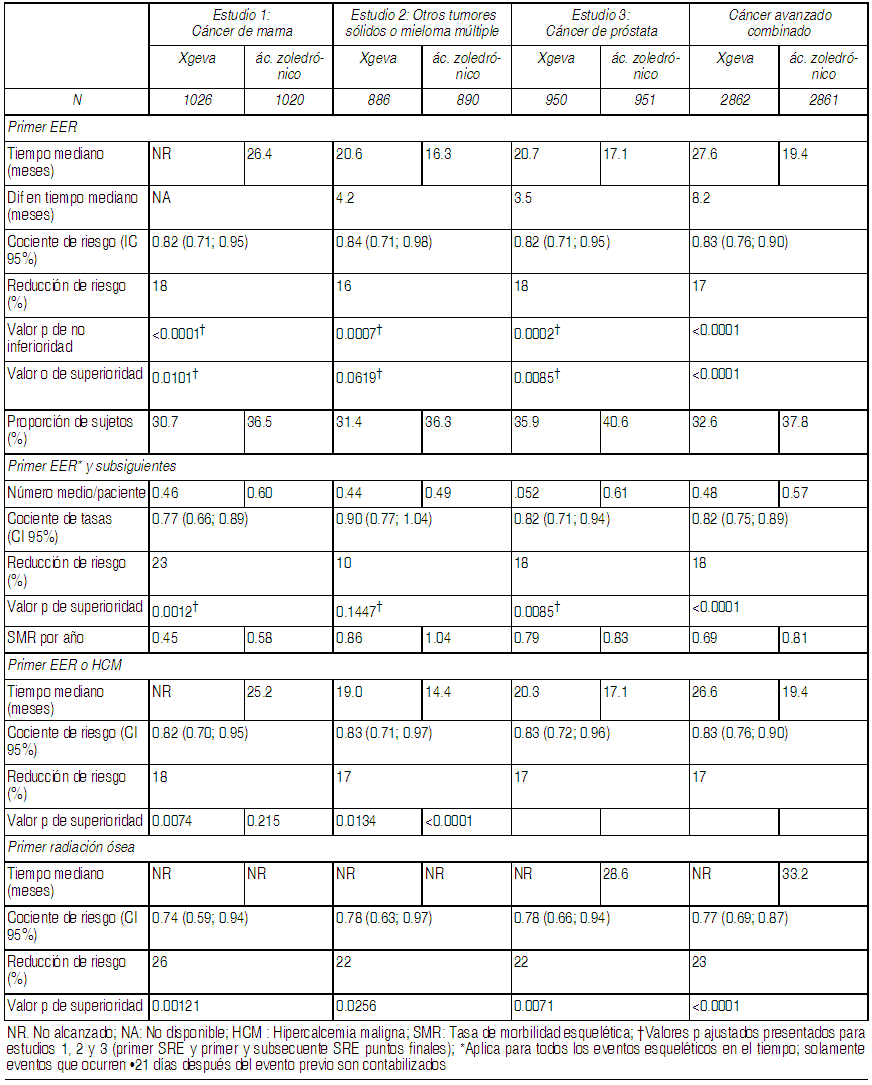
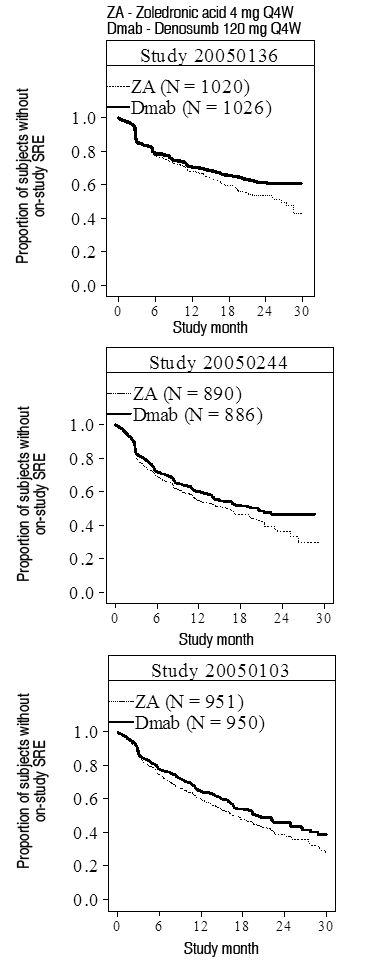
Farmacodinamia: Mecanismo de acción: Denosumab es un anticuerpo monoclonal humano (IgG2) que se dirige y fija con gran afinidad y especificidad al RANKL, evitando que éste active su único receptor, el RANK, el cual se encuentra en la superficie de los osteoclastos y sus precursores. El ligando de RANK existe como una proteína transmembrana o en forma soluble. El ligando de RANK es esencial en la formación, función y supervivencia de los osteoclastos, el único tipo celular responsable de la resorción ósea. El incremento en la actividad de los osteoclastos, estimulada por el ligando de RANK, es un mediador clave de la destrucción ósea en la osteopatía en tumores metastásicos y mieloma múltiple. La prevención de la interacción de RANK-RANKL ocasiona una reducción en el número de osteoclastos y su función, disminuyendo así la resorción ósea y la destrucción ósea inducida por cáncer. Efectos farmacodinámicos: En un estudio de fase 2 realizado en pacientes con cáncer de mama y metástasis óseas que no habían recibido previamente terapia con bisfosfonatos IV, las dosis SC de 120 mg de Xgeva cada 4 semanas ocasionaron una rápida reducción en los marcadores de resorción ósea (uNTX/creatinina y CTx en suero), con una reducción mediana de 82% en uNTX/Cr en un plazo de 1 semana. Se mantuvieron las reducciones en los marcadores de recambio óseo, con reducciones medianas de uNTX/Cr de 74% a 82% desde la semana 2 a la 25 de una dosificación continua de 120 mg cada 4 semanas. En estudios de fase 3 de pacientes con cáncer avanzado, se observaron reducciones medianas de aproximadamente 80% en la relación uNTx/Cr respecto al basal después de 3 meses de tratamiento entre 2.075 pacientes con cáncer avanzado (mama, próstata, mieloma múltiple u otros tumores sólidos) tratados con Xgeva. De manera similar, en pacientes con cáncer avanzado y metástasis óseas (incluyendo sujetos con mieloma múltiple y osteopatías) que estaban recibiendo terapia con bisfosfonatos IV, y que aún mostraban niveles de uNTX/Cr > 50 nM/mM, la dosificación SC múltiple de Xgeva, administrado ya sea cada 4 semanas o cada 12 semanas, ocasionó una reducción aproximada de 80% en la relación uNTX/creatinina respecto al basal después de 3 y 6 meses de tratamiento. En general, 97% de los pacientes en los grupos de tratamiento con Xgeva tuvieron cuando menos 1 valor de uNTX/Cr < 50 nM/mM hasta la semana 25 del estudio. Inmunogenicidad: En estudios clínicos, no se han observado anticuerpos neutralizantes para XGEVA. Utilizando un inmunoensayo sensible, <1% de los pacientes tratados con denosumab arrojaron resultados positivos respecto a anticuerpos de unión no neutralizantes, sin indicios de alteraciones en las respuestas farmacocinética, farmacodinámica o en el perfil de toxicidad. Farmacocinética: Después de la administración subcutánea, la biodisponibilidad fue de 62% y denosumab mostró un perfil farmacocinético no lineal con dosis superiores a un amplio intervalo de dosificación, pero con incrementos aproximadamente proporcionales a las dosis en los niveles de exposición a dosis de 60 mg (ó 1 mg/kg) y superiores. Al administrar dosis múltiples de 120 mg cada 4 semanas, se observó una acumulación aproximada del doble en las concentraciones séricas de denosumab, y el estado estable se alcanzó a los 6 meses, lo cual es consistente con el perfil farmacocinético independiente del tiempo. En el estado estable, la concentración media mínima en el suero fue de 20.6 mcg/ml (intervalo: 0.456 a 56.9 mcg/ml). En sujetos que suspendieron la dosificación de 120 mg cada 4 semanas, la vida media promedio fue de 28 días (intervalo: 14 a 55 días). Se realizó un análisis farmacocinético poblacional para evaluar los efectos de las características demográficas. Este análisis no mostró diferencias notables en el perfil farmacocinético respecto a la edad (18 a 87 ańos), raza, peso corporal (36 a 174 kg), o entre los pacientes con tumores sólidos. Los perfiles farmacocinético y farmacodinámico del denosumab fueron similares en hombres y en mujeres, así como en pacientes transferidos de una terapia con bisfosfonatos IV. Poblaciones de pacientes especiales: Pacientes de edad avanzada (de 65 ańos de edad o mayores): En un análisis farmacocinético poblacional de pacientes cuyas edades variaron de los 18 a los 87 ańos de edad, no se encontró que la edad fuera un factor que afectara la farmacocinética del denosumab. Nińos y adolescentes (de hasta 18 ańos de edad): No se dispone de datos farmacocinéticos en pacientes pediátricos (véase Datos preclínicos de seguridad). Raza: La farmacocinética del denosumab no se vio afectada por la raza en pacientes tratados con denosumab. Insuficiencia renal: En un estudio realizado en 55 pacientes sin cáncer avanzado pero con grados variables de función renal, incluyendo pacientes sometidos a diálisis, el grado de insuficiencia renal no produjo efecto alguno en la farmacocinética y la farmacodinamia del denosumab; por lo tanto, no es necesario ajustar la dosis en presencia de insuficiencia renal. Los pacientes con insuficiencia renal grave (depuración de creatinina <30 ml/min) o sometidos a diálisis se encuentran en mayor riesgo de desarrollar hipocalcemia. Insuficiencia hepática: No se han realizado estudios clínicos para evaluar el efecto de la insuficiencia hepática en la farmacocinética del denosumab. Estudios clínicos: Prevención de eventos esqueléticos relacionados en adultos con neoplasia maligna avanzada que compromete hueso: Se evaluaron los perfiles de eficacia y seguridad de Xgeva en la prevención de Eventos Esqueléticos Relacionados (EERs) en pacientes con cáncer avanzado y lesiones óseas en 3 estudios aleatorizados, doble ciegos y controlados con activo. Cada estudio evaluó denosumab (120 mg administrados vía subcutánea) con ácido zoledrónico (4 mg administrados vía intravenosa, con ajuste de dosis para función renal disminuida) 1 vez cada 4 semanas. Los criterios primarios y secundarios de valoración evaluaron la ocurrencia de uno o más EERs definidos como cualquiera de lo siguiente: fractura patológica, radioterapia ósea, cirugía ósea o compresión de la médula espinal. Xgeva redujo o previno el riesgo de desarrollar un EER, o de desarrollar EERs múltiples (primero o subsiguiente) en pacientes con cáncer avanzado que compromete hueso. Los resultados de eficacia se presentan en la Tabla 1.
Ver Tabla
Ver Tabla
Efecto en el dolor: Los análisis del dolor incluyeron la evaluación de los cambios respecto al basal en la calificación del peor dolor según la BPI-SF; las evaluaciones del tiempo hasta el agravamiento del dolor, dolor moderado o grave o mejoría en el dolor; y las proporciones de sujetos que satisficieron estos criterios. En un análisis ad-hoc del conjunto de datos combinados, el tiempo mediano hasta el agravamiento del dolor (calificación del peor dolor >4 puntos e incremento respecto al basal de ł 2 puntos) fue más prolongado para Xgeva en comparación con el ácido zoledrónico (65 contra 59 días y 181 contra 169 días, respectivamente). En un análisis adicional ad hoc de subgrupos de pacientes con dolor leve o sin dolor en la línea basal, el tiempo hasta el agravamiento del dolor (calificación del peor dolor >4 puntos) sufrió una demora en el grupo tratado con Xgeva, en comparación con el grupo de tratamiento con ácido zoledrónico (198 contra 143 días). El tiempo hasta la mejoría en el dolor (por ejemplo, disminución respecto al basal de ł 2 puntos en la calificación del peor dolor según la BPI-SF) fue similar para denosumab y para el ácido zoledrónico en cada estudio y en los análisis integrados. Sobrevida global y progresión de la enfermedad: La progresión de la enfermedad fue similar entre Xgeva y el ácido zoledrónico en los 3 estudios y en el análisis preespecificados de los 3 estudios combinados. En los 3 estudios, la sobrevida global entre Xgeva y el ácido zoledrónico en pacientes con cáncer avanzado que compromete hueso fue balanceada: pacientes con cáncer de mama (el cociente de riesgo y el IC 95% fue de 0.95 [0.81; 1.11]), pacientes con cáncer de próstata (el cociente de riesgo y el IC 95% fue de 1.03 [0.91, 1.17]), y pacientes con otros tumores sólidos o mieloma múltiple (el cociente de riesgo y el IC 95% fue de 0.95 [0.83; 1.08]). Un análisis ad hoc en el estudio 2 (pacientes con otros tumores sólidos o mieloma múltiple) examinó la sobrevida global para a los 3 tipos de tumores utilizados en la estratificación (cáncer pulmonar de células no pequeńas, mieloma múltiple y otro). La sobrevida global fue más prolongada para XGEVA en pacientes con cáncer pulmonar de células no pequeńas (cociente de riesgo [IC 95%] de 0.79 [0.65; 0.95]; n= 702) y más prolongada para el ácido zoledrónico en pacientes con mieloma múltiple (cociente de riesgo [IC 95%] de 2.26 [1.13; 4.50]; n= 180), y similar entre Xgeva y el ácido zoledrónico en otros tipos de tumores (cociente de riesgo [IC 95%] de 1.08 (0.90; 1.30); n= 894). Este estudio no controló para factores pronósticos ni tratamientos antineoplásicos de mieloma multiple. En un análisis preespecificado combinado de los estudios 1, 2 y 3, la sobrevida global fue similar entre Xgeva y el ácido zoledrónico (cociente de riesgo e IC 95% de 0.99 [0.91; 1.07]). Datos preclínicos de seguridad: Carcinogenicidad: No se ha evaluado el potencial carcinogénico del denosumab en estudios a largo plazo realizados en animales. Mutagenicidad: No se ha evaluado el potencial genotóxico del denosumab. Toxicología en la reproducción: Fertilidad: El denosumab no produjo efecto alguno en la fertilidad femenina ni en los órganos reproductivos masculinos en monos sometidos a niveles de exposición que fueron 9.5 a 16 veces superiores, respectivamente, al nivel de exposición en seres humanos a 120 mg SC administrados 1 vez cada 4 semanas. Farmacología en animales: Se ha demostrado que el denosumab es un potente inhibidor de la resorción ósea a través de la inhibición del ligando de RANK. Como la actividad biológica del denosumab en animales es específica para los primates no humanos, se utilizó la evaluación de ratones modificados genéticamente (genes inactivados), o el uso de otros inhibidores biológicos de la vía RANK/RANKL, como OPG-Fc y RANK-Fc, para evaluar las propiedades farmacodinámicas del denosumab en modelos de roedores. En modelos de ratones con metástasis ósea para cáncer pulmonar de células no pequeńas, cáncer de próstata y cáncer de mama humanos positivos y negativos a receptores de estrógenos, el OPG-Fc redujo las lesiones osteolíticas, osteoblásticas y osteolíticas/osteoblásticas, retrasó la formación de metástasis óseas de novo y redujo el crecimiento tumoral esquelético. Hubo una inhibición adicional en el crecimiento tumoral esquelético al combinar OPG-Fc con terapia hormonal (tamoxifeno) en un modelo de cáncer de mama, o con quimioterapia (docetaxel) en modelos de cáncer de próstata y cáncer pulmonar. En un modelo de ratón de inducción tumoral mamaria, el RANK-Fc retrasó la formación de tumores. Primates adolescentes que recibieron dosis de denosumab 15 veces (dosis de 50 mg/kg) y 2.8 veces (dosis de 10 mg/kg) la exposición del área bajo la curva (AUC) en humanos adultos tratados con 120 mg por vía subcutánea cada 4 semanas, tuvieron placas de crecimiento anormal, las cuales se consideraron consistentes con la actividad farmacológica del denosumab. En monos cynomolgus recién nacidos expuestos in utero a denosumab a razón de 50 mg/kg, hubo mortalidad postnatal; un crecimiento óseo anormal que provocó una fuerza ósea reducida, disminución de la hematopoyesis, malformación dental; ausencia de nódulos linfáticos periféricos; y disminución del crecimiento neonatal. Después de un período de recuperación desde el nacimiento hasta los 6 meses de edad, los efectos en los huesos regresaron a la normalidad; no hubo efectos adversos en la erupción dentaria; y se observó mínima a moderada mineralización en múltiples tejidos en un animal recuperado. El desarrollo de las glándulas mamarias maternas fue normal. Los ratones noqueados que carecen de RANK o RANKL (1) tuvieron una ausencia de lactancia debido a la inhibición de la maduración de las glándulas mamarias (desarrollo de la glándula lobuloalveolar durante el embarazo) (2) exhibieron un deterioro en la formación de ganglios linfáticos (3) exhibieron una reducción en el crecimiento óseo, una alteración en las placas de crecimiento y una falta de dentición. Se observaron casos de reducción en el crecimiento ósea, alteración en las placas de crecimiento y deterioro en la dentición en estudios realizados en ratas recién nacidas que recibieron inhibidores de RANKL, y estos cambios fueron parcialmente reversibles al suspender la dosificación de inhibidores de RANKL (véase Embarazo y lactancia). Los estudios de distribución histológica indicaron que el denosumab no se une a tejidos conocidos por expresar a otros miembros de la superfamilia de TNF, incluyendo el ligando inductor de apoptosis relacionado al TNF (TRAIL, por sus siglas en inglés).
|
|
Posología:
Administración: Debe ser administrada por una persona entrenada en la técnica de aplicación de inyecciones. Dosis: La dosis recomendada de Xgeva consiste en una inyección subcutánea de 120 mg, administrada 1 vez cada 4 semanas en muslos, abdomen o antebrazos. Los pacientes deben recibir complementos de calcio y vitamina D mientras se encuentren bajo tratamiento. Poblaciones: Nińos: Xgeva no se recomienda en pacientes pediátricos, ya que aún no se han establecido los perfiles de seguridad y eficacia de Xgeva en ese grupo de edad. En estudios realizados en animales, se ha acoplado la inhibición del ligando RANK/RANK (RANKL), mediante un constructo de osteoprotegerina unido a Fc (OPG-Fc), a la inhibición del crecimiento óseo y la falta de erupción de dientes (véase Datos preclínicos de seguridad). Por lo tanto, el tratamiento con denosumab es capaz de afectar el crecimiento de los huesos en nińos con placas epifisiarias abiertas, y puede inhibir la erupción de los dientes. Pacientes de edad avanzada: No se observaron diferencias en la seguridad o la eficacia entre pacientes mayores y pacientes jóvenes. De acuerdo a los datos disponibles sobre la seguridad y la eficacia en pacientes de edad avanzada, no se requiere ajuste de dosis (véase Farmacocinética: Poblaciones de pacientes especiales). Insuficiencia renal: De acuerdo a los datos disponibles sobre la seguridad y la eficacia, no se requiere ajustar la dosis en pacientes con insuficiencia renal y no es necesario el monitoreo renal cuando reciben Xgeva (véase Farmacocinética: Poblaciones de pacientes especiales). Los pacientes con insuficiencia renal grave (depuración de creatinina <30 ml/min) o sometidos a diálisis se encuentran en mayor riesgo de desarrollar hipocalcemia. Es importante que se instituya una ingesta adecuada de calcio y vitamina D en pacientes con insuficiencia renal grave o que reciban diálisis. Insuficiencia hepática: Aún no se han estudiado los perfiles de seguridad y eficacia de Xgeva en pacientes con insuficiencia hepática.
|
|
Modo de Empleo:
Instrucciones para su uso/manejo: Antes de su administración, se debe inspeccionar la solución de Xgeva en cuanto a presencia de partículas y decoloración. La solución no debe utilizarse si se encuentra turbia o decolorada. No agitar. Se recomienda utilizar una aguja de calibre 27 para la administración de denosumab. No volver a introducir el frasco vial. Se deberá desechar cualquier producto no utilizado o material de desecho, de conformidad con los requisitos locales. No todas las presentaciones se encuentran disponibles en todos los países.
|
|
Efectos Colaterales:
Datos de estudios clínicos: A continuación se enlistan las reacciones adversas por clase de sistema de órganos corporales según MedDRA, así como por frecuencia. A continuación se presentan las categorías de frecuencia basadas en tasas de eventos ocurridos durante un ańo: Muy común: ł 1 en 10. Común: ł 1 en 100 y <1 en 10. No común: ł 1 en 1000 y <1 en 100. Rara: ł 1 en 10000 y <1 en 1000. Muy rara: <1/10000. Dentro de cada agrupación de frecuencia y clase de sistema de órganos, se presentan los efectos adversos en orden de severidad decreciente. Clase de sistema de órganos según MedDRA: Categoría de frecuencia: Efecto adverso. Trastornos del sistema inmune: Raro: Hipersensibilidad al medicamento. Trastornos metabólicos y nutricionales: Común: Hipocalcemia1,2. Común: Hipofostatemia. Trastornos respiratorios, torácicos y mediastínicos: Muy común: Disnea. Trastornos musculoesqueléticos y del tejido conjuntivo: Común. Osteonecrosis mandibular1. Raro: Fractura femoral atípica. 1 Véase Advertencias y Precauciones. 2 Datos de postmercadeo. Osteonecrosis mandibular (ONJ): En 3 estudios clínicos fase III activo-controlado en pacientes con neoplasias avanzadas que involucran hueso, se confirmó ONJ en 1.8% de los pacientes en el grupo con Xgeva (mediana de exposición de 12.0 meses; rango 0.1 - 40.5) y 1.3% de los pacientes en el grupo con ácido zoledrónico. Los estudios en pacientes con cáncer de mama o próstata incluyeron una extensión en la fase de tratamiento con Xgeva (mediana de exposición general de 14.9 meses; rango 0.1-67.2). La incidencia ajustada paciente ańo de ONJ confirmado fue de 1.1% durante el primer ańo de tratamiento y de 4.1% de ahí en adelante. La mediana de tiempo para la ONJ fue de 20.6 meses (rango: 4-53). Datos de postmercadeo: Hipocalcemia grave: Se han reportado casos graves, e incluso fatales de hipocalcemia síntomática. Reacciones de hipersensibilidad: Reacciones de hipersensibilidad, incluyendo anafilaxia.
|
|
Contraindicaciones:
Hipocalcemia, hipersensibilidad clínicamente significativa a denosumab o cualquiera de los componentes de Xgeva.
|
|
Advertencias:
Hipocalcemia: En estudios clínicos realizados en pacientes con cáncer avanzado tratado con Xgeva o ácido zoledrónico, con mayor frecuencia se reportaron casos de hipocalcemia en el grupo tratado con denosumab (9.6%), en comparación con el grupo que recibió ácido zoledrónico (5.0%). Además, con mayor frecuencia se observó disminución grado 3 y grado 4 de los niveles de calcio en suero en pacientes que recibieron denosumab en comparación con el grupo que recibió ácido zoledrónico. La hipocalcemia preexistente se debe corregir antes de iniciar el tratamiento con Xgeva. Se requiere una complementación de calcio y vitamina D en todos los pacientes, a menos que se presente hipercalcemia. Si se presenta hipocalcemia, es posible que se requiera una complementación adicional a corto plazo de calcio (véase Efectos colaterales). Osteonecrosis mandibular (ONM): Se confirmaron casos de osteonecrosis mandibular (ONM) en el 1.8% de los pacientes tratados con Xgeva y un 1.3% en los pacientes tratados con ácido zoledronico (Véase Efectos colaterales). En estudios clínicos, la incidencia de ONM fue más alta con exposiciones mas prolongadas. El prescriptor debe realizar una exploración oral antes de iniciar el tratamiento con Xgeva, y se debe contemplar una exploración dental con cuidados dentales preventivos adecuados antes de iniciar el tratamiento con Xgeva en pacientes que tengan factores de riesgo de desarrollar ONM. Se deben mantener buenas prácticas de higiene bucal durante el tratamiento con Xgeva. Mientras se encuentren bajo tratamiento, los pacientes deberán evitar en la medida de lo posible procedimientos dentales invasivos. En aquellos pacientes que requieran procedimientos dentales invasivos, el juicio clínico del médico tratante y/o el cirujano maxilofacial deberán guiar el manejo de cada paciente. Los pacientes con sospecha o diagnóstico de ONM mientras se encuentren bajo tratamiento con Xgeva deberán recibir cuidados de un dentista o cirujano bucal. Fractura femoral atípica: Se ha reportado fractura femoral atípica con Xgeva. Con traumatismo leve o sin él, puede ocurrir fractura femoral atípica en la región subtrocanterea y diáfisis del fémur y puede ser bilateral. Los hallazgos radiográficos caracterizan estos eventos. También se ha reportado fractura femoral atípica en pacientes con ciertas condiciones comórbidas (por ej., deficiencia de vitamina D, artritis reumatoide, hipofosfatasia) y con el uso de ciertos agentes farmacéuticos (por ej., bifosfonatos, glucocorticoides, inhibidores de la bomba de protones). También han ocurrido estos eventos sin terapia anti-resorción. Durante el tratamiento con Xgeva, debe aconsejarse a los pacientes que reporte cualquier dolor inusual en muslos, cadera, o ingle. Los pacientes que presenten dichos síntomas deben ser evaluados para buscar alguna fractura femoral incompleta, y también debe examinarse el fémur contralateral. Fármacos con el mismo ingrediente activo: Xgeva contiene el mismo ingrediente activo que se encuentra en Prolia (denosumab). Los pacientes bajo tratamiento con Xgeva no deben recibir Prolia.
|
|
Precauciones:
Embarazo y lactancia: Embarazo: Xgeva puede causar dańo fetal cuando se administra a mujeres embarazadas. Con base en los hallazgos en animales, se espera que Xgeva tenga efectos reproductivos adversos. La exposición en el útero de denosumab a monos Cynomologus dieron lugar a un mayor número de pérdidas fetales, mortinatos y mortalidad postnatal, junto con una evidencia de ausencia de ganglios linfáticos periféricos, el crecimiento anormal de huesos y disminución del crecimiento neonatal. No hay estudios adecuados y bien controlados con Xgeva en mujeres embarazadas. Las mujeres deben ser advertidas de no quedar embarazadas cuando toman Xgeva. Si se utiliza este fármaco durante el embarazo o si la paciente quedara embarazada mientras toma este medicamento, se debe informar a la paciente del potencial dańo para el feto. En otro estudio con monos Cynomolgus a los que se administró denosumab durante todo el embarazo a exposiciones AUC hasta 12 veces más altas que la dosis humana (120 mg cada 4 semanas), hubo aumento de muertes fetales y postnatales; el crecimiento óseo anormal resultó en una fuerza ósea reducida, disminución de la hematopoyesis, y malformación dental; ausencia de nódulos linfáticos periféricos; y disminución del crecimiento neonatal. No hubo evidencia de dańo materno antes del parto; efectos adversos maternos se presentaron infrecuentemente durante el parto. El desarrollo de las glándulas mamarias maternas fue normal. Los estudios realizados en ratones con genes inactivados (Knockout) sugieren que la ausencia del RANKL podría interferir con la maduración de las glándulas mamarias maternas, conduciendo así a una lactancia posparto alterada. A las mujeres que se embaracen durante el tratamiento con Xgeva se les alienta a participar en el Programa de Vigilancia de Embarazos de Amgen. Las pacientes o sus médicos deberán contactar al representante local de GSK para el reclutamiento. Lactancia: Se desconoce si denosumab se excreta en la leche humana. Como denosumab tiene el potencial de ocasionar reacciones adversas en lactantes amamantados, se deberá elegir entre suspender la lactancia materna o suspender la administración del medicamento. Efectos en la capacidad de conducir y utilizar maquinaria: No se han realizado estudios sobre el efecto en la capacidad de conducir o utilizar maquinaria pesada en pacientes que reciben tratamiento con denosumab.
|
|
Interacciones Medicamentosas:
No se han realizado estudios sobre interacciones medicamentosas con Xgeva. En estudios clínicos, Xgeva ha sido administrado en combinación con tratamiento antineoplásico estándar y en sujetos que recibieron previamente bisfosfonatos. Los perfiles farmacocinético y farmacodinámico de denosumab no se vieron alterados por la quimioterapia y/o terapia hormonal concomitante, ni por la exposición previa a bisfosfonatos intravenosos.
|
|
Sobredosificación:
No se dispone de datos obtenidos de estudios clínicos en humanos en relación con la sobredosificación.
|
|
Incompatibilidades:
Este medicamento no debe mezclarse con otros medicamentos.
|
|
Conservación:
Vida de anaquel: La fecha de caducidad se indica en el empaque. Precauciones especiales de almacenamiento: Almacenar en un refrigerador (2ş C-8 şC). No congelar. Conservar el frasco vial de vidrio en la caja externa a fin de protegerla de la luz directa. No agitar. Si se retira de refrigeración, Xgeva debe conservarse a una temperatura ambiente controlada (hasta 25ş C [77 şF]) en su empaque de cartulina original, y deberá utilizarse dentro de los 30 días siguientes.
|
|
Presentaciones:
Naturaleza y contenido del empaque: Xgeva es un producto estéril y libre de conservadores. Solución de 1.7 ml (70 mg/ml) en un vial para uso único fabricado con vidrio tipo I.
|
|



{kind=link}
{kind=link}