 |
Composición:
Taxotere 20 mg: cada vial de Taxotere 20 mg contiene: Docetaxel Trihidrato 21.34 mg, equivalente a 20 mg de docetaxel (anhidro) en 0.5 ml de Polisorbato 80 (con un volumen de llenado 24.4 mg/0.61ml). Se ha establecido este volumen para compensar la pérdida de líquido durante la preparación de la premezcla, debido a la espuma, adhesión a las paredes del vial y al "volumen muerto". Este sobrellenado asegura que después de la dilución con el contenido completo del vial de solvente adjunto, el volumen mínimo de pre-mezcla extraíble sea de 2 ml. Solvente para Taxotere 20 mg: cada vial de solvente para Taxotere 20 mg contiene 1.5 ml (1.98 volumen final de llenado). La adición del contenido del vial de solvente completo al contenido del vial de Taxotere 20 mg, asegura una pre-mezcla a una concentración de docetaxel de 10mg/ml. Taxotere 80 mg: cada vial de Taxotere 80 mg contiene: Docetaxel Trihidrato 85.36 mg, equivalente a 80 mg de Docetaxel (anhidro) en 2 ml de Polisorbato 80 (con un volumen de llenado 94.4 mg/2.36 ml). Se ha establecido este volumen para compensar la pérdida de líquido durante la preparación de la pre-mezcla, debido a la espuma, adhesión a las paredes del vial y al "volumen muerto". Este sobrellenado asegura que después de la dilución con el contenido completo del vial de solvente adjunto, el volumen mínimo de premezcla extraíble sea de 8 ml. Solvente para Taxotere 80 mg: cada vial de solvente para Taxotere 80 mg contiene 6 ml (7.33 volumen final de llenado). La adición del contenido del vial de solvente completo al contenido del vial de Taxotere 80 mg asegura una pre-mezcla a una concentración de docetaxel de 10 mg/ml.
|
|
Acción Terapéutica:
Grupo fármacoterapéutico: Agentes antineoplásicos.
|
|
Indicaciones:
Adyuvancia: Taxotere® en combinación con doxorrubicina y ciclofosfamida, está indicado para el tratamiento adyuvante de pacientes con cáncer de mama operable, linfonodo positivo. Doxorrubicina y ciclofosfamida seguidos de Taxotere® en combinación con trastuzumab (ACTH) está indicado para el tratamiento adyuvante de pacientes con cáncer de mama operable cuyos tumores sobreexpresen HER 2. Taxotere® en combinación con trastuzumab y carboplatino (TCH) está indicado para el tratamiento adyuvante de pacientes con cáncer de mama operable cuyos tumores sobreexpresen HER 2. Metastático: Taxotere® en combinación con doxorrubicina está indicado para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastático, que no han recibido previamente terapia citotóxica para esta condición. Taxotere® en combinación con trastuzumab, está indicado para el tratamiento de pacientes con cáncer de mama cuyo tumor sobreexprese el HER 2 y que no han recibido quimioterapia previa para enfermedad metastásica. La monoterapia con Taxotere® está indicada para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastático después del fracaso de la quimioterapia de primera línea o subsecuentes. La quimioterapia previa deberá haber incluido una antraciclina o un agente alquilante. Taxotere® en combinación con capecitabina está indicado para el tratamiento de pacientes con cáncer de mama localmente avanzado o metastático después del fracaso a quimioterapia citotóxica que haya incluido una antraciclina. Cáncer de pulmón de células no-pequeńas. Cáncer de ovario metastásico, después del fracaso de la quimioterapia de primera línea o subsecuentes. Cáncer de cabeza y cuello. Taxotere® en combinación con cisplatino y 5fluoruracilo, está indicado para el tratamiento en aquellos pacientes con carcinoma de células escamosas locamente avanzado de cabeza y cuello, no resecable (estadio III y IV). Cáncer de próstata: Taxotere en combinación con prednisona o prednisolona está indicado para el tratamiento de pacientes con cáncer de próstata metastático andrógeno independiente (hormonorefractario). Adenocarcinoma gástrico: Taxotere® en combinación con cisplatino y 5fluorouracilo está indicado para el tratamiento de pacientes con adenocarcinoma gástrico avanzado, incluyendo el adenocarcinoma de la unión gastroesofágica, que no hayan recibido quimioterapia previa para cáncer gástrico avanzado.
|
|
Propiedades:
Propiedades farmacodinámicas: Información preclínica. Docetaxel es un agente antineoplásico que actúa estimulando el ensamblaje de tubulina en microtúbulos estables e inhibe su desintegración, lo que lleva a una notable disminución de tubulina libre. La ligadura de docetaxel a microtúbulos no altera la cantidad de protofilamentos. Se ha demostrado que docetaxel in vitro altera la red microtubular en las células, la que es vital para las funciones celulares en mitosis y en interfase. In vitro, se demostró que docetaxel es citotóxico frente a varias líneas celulares de tumores murinos y humanos y frente a células tumorales humanas recién obtenidas de ensayos de clonación. Docetaxel alcanza altas concentraciones intracelulares en un prolongado tiempo de permanencia celular. Docetaxel es activo en algunas pero no en todas las líneas celulares que sobreexpresan la pglicoproteína, la que es codificada por un gen de multirresistencia. In vivo, docetaxel es un agente no catalogable y posee un amplio espectro de actividad antitumoral experimental contra injertos tumorales murinos y humanos avanzados. Farmacocinética: Las propiedades farmacocinéticas de docetaxel fueron evaluadas en pacientes con cáncer luego de la administración de 20115 mg/m2 en estudios de Fase I. El perfil farmacocinético de docetaxel es dosisindependiente y coherente con un modelo farmacocinético de tres compartimentos con vida medias para las fases a , ß y g de 4 min., 36 min. y 11.1 h, respectivamente. La fase tardía se debe, en parte, a un efluvio relativamente lento de docetaxel desde el compartimiento periférico. Tras la administración de una dosis de 100 mg/m2 en forma de infusión de 1 hora, se obtiene un pico medio de nivel plasmático de 3.7 µg/ml con una AUC correspondiente de 4.6 h*µg/ml. Los valores medios para el total de depuración corporal y volumen de distribución a equilibrio dinámico de 21 l/h/m2 y 113 l, respectivamente. La variación interindividual en el total de depuración corporal fue aproximadamente 50%. Docetaxel se une en más del 95% a las proteínas plasmáticas. Un estudio 14Cdocetaxel fue efectuado en tres pacientes con cáncer. Docetaxel se eliminó tanto en orina como en heces luego de un metabolismo oxidativo del grupo éster terbutilo mediado por el citocromo P450. En 7 días, la excreción urinaria y fecal fue de aproximadamente 6% a 75% de la radioactividad administrada, respectivamente. Alrededor del 80% de la radioactividad recuperada en las heces se excretó durante las primeras 48 horas en forma de un metabolismo inactivo principal y 3 secundarios y muy pocas cantidades de droga inalterada. Se realizó un análisis farmacocinético con docetaxel en una población de 577 pacientes. Los parámetros farmacocinéticos estimados por el modelo resultaron muy cercanos a aquellos estimados a partir de los estudios de Fase. I. No se observaron alteraciones farmacocinéticas para docetaxel conforme a la edad o sexo del paciente. En una reducida cantidad de pacientes (n=23), con datos químicos clínicos que sugerían una insuficiencia de función hepática entre leve a moderada (ALT, AST ł 1.5 veces el límite superior normal [ULN] asociado con fosfatasa alcalina ł 2.5 veces el límite superior normal ULN), se observó una disminución del 27% de clearance, como promedio. El clearance de docetaxel no se modificó en pacientes con retención de líquidos leve a moderada y no hay información disponible en pacientes con retención grave de líquidos. Cuando se lo usa en terapia combinada, docetaxel no ejerce influencia sobre la depuración de doxorubicina y los niveles plasmáticos de doxorrubicinol (un metabolito de doxorubicina). Además, aumentó la depuración de docetaxel mientras se mantuvo la eficacia. La farmacocinética de docetaxel, doxorubicina y ciclofosfamida, fue estudiada, en 30 pacientes con cáncer de mama, no hubo influencia por la co administración. Estudios fase I evaluando el efecto de capecitabina sobre la farmacocinética de docetaxel, y el efecto de docetaxel en la farmacocinética de capecitabina mostró que no hubo efecto de capecitabina sobre docetaxel (Cmax y AUC) y no hubo efecto de docetaxel en la farmacocinética de 5'DFUR (el metabolito más importante de capecitabina). La depuración de docetaxel en terapia combinada con cisplatino o carboplatino fue similar a la observada luego de monoterapia con docetaxel. El perfil farmacocinético de cisplatino administrado enseguida luego de la infusión de Taxotere es similar al observado con cisplatino solo. En 42 pacientes se estudió el efecto de prednisona sobre la farmacocinética de docetaxel administrado con premedicación estándar de dexametasona. No se observó ningún de la prednisona sobre la farmacocinética de docetaxel. La administración combinada de docetaxel, cisplatino y 5fluoruracilo en 12 pacientes con tumores sólidos no influenció la farmacocinética de cada droga en forma individual. Datos preclínicos sobre seguridad: Carcinogénesis: No se ha estudiado el potencial carcinogénico de docetaxel. Mutagénesis: Docetaxel ha demostrado ser mutagénico en la prueba de micronúcleo y aberración cromosómica in vitro en células CHOK1 y en la prueba de micronúcleo in vivo en ratones. Sin embargo, no indujo mutagenicidad en la prueba Ames o en la prueba CHO/HGPRT de mutación de gen. Estos resultados son coherentes con la actividad farmacológica de docetaxel. Dańo en la fertilidad: Los efectos adversos sobre los testículos observados en estudios de toxicidad con roedores sugieren que docetaxel puede dańar la fertilidad masculina. Características farmacéuticas: Vida útil: Taxotere 20 mg en viales: 24 meses Taxotere 80 mg en viales: 36 meses.
|
|
Posología:
El uso de docetaxel se debe restringir a unidades especializadas en la administración de quimioterapia citotóxica y debe ser administrado bajo la supervisión de un médico experto en el uso de quimioterapia antineoplásica. Premedicación: debe usarse una premedicación consistente en corticoesteroides orales, tales como dexametasona 16 mg por día (por ej: 8 mg, 2 veces al día) durante 3 días, empezando el día antes de la administración de docetaxel, salvo contraindicación. Para el cáncer de próstata, en virtud de prednisona o prednisolona concomitante, el régimen recomendado de premedicación es dexametasona oral 8 mg, 12 horas, 3 horas y 1 hora antes de la infusión de docetaxel. Se puede utilizar G-CSF profiláctico para disminuir el riesgo de toxicidad hematológica. Administración: docetaxel es administrado como infusión de 1 hora cada 3 semanas. Dosis recomendadas: Cáncer de mama: En adyuvancia: en el tratamiento de adyuvancia en pacientes con cáncer de mama operable linfonodo positivo, la dosis recomendada de docetaxel es 75 mg/m2 administrado 1 hora después 50 mg/m2 de doxorubicina y de 500 mg/m2 de ciclofosfamida, cada 3 semanas durante 6 ciclos. Para el tratamiento de adyuvancia de pacientes con cáncer de mama operable cuyos tumores se sobreexpresen HER2: AC-TH: AC (ciclos 1 - 4): doxorubicina (A) 60 mg/m2 seguido de ciclofosfamida (C) 600 mg/m2 administrado cada 3 semanas por 4 ciclos. TH (ciclos 5 - 8): docetaxel (T) 100 mg/m2 administrado cada 3 semanas por 4 ciclos, y trastuzumab (H) administrado semanalmente de acuerdo al siguiente esquema: Ciclo 5 (comenzando 3 semanas despues del ultimo ciclo de AC): Dia 1: trastuzumab 4 mg/kg (dosis de carga). Dia 2: docetaxel 100 mg/m2. Dias 8 y 15: trastuzumab 2 mg/kg. Ciclos 6 - 8: Dia 1: docetaxel 100 mg/m2 y trastuzumab 2 mg/kg: Dias 8 y 15: trastuzumab 2 mg/kg. 3 semanas después del día 1 del ciclo 8: trastuzumab 6 mg/kg administrado cada 3 semanas. Trastuzumab es administrado por una duración total de 1 ańo. TCH: TCH (ciclos 1 - 6): docetaxel (T) 75 mg/m2 y carboplatino (C) a AUC de 6 mg/ml/min administrado cada 3 semanas y trastuzumab (H) administrado semanalmente de acuerdo con el siguiente esquema: Ciclo 1: Día 1: trastuzumab 4 mg/kg (dosis de carga). Día 2: docetaxel 75 mg/m2 y carboplatino a AUC de 6 mg/ml/min. Días 8 y 15: trastuzumab 2 mg/kg. Ciclos 2 - 6: Día 1: docetaxel 75 mg/m2 seguido de carboplatino a AUC de 6 mg/ml/min y trastuzumab 2 mg/kg. Días 8 y 15: trastuzumab 2 mg/kg. Metastático: En tratamiento de primera linea, docetaxel 75 mg/m2 de Taxotere R mas trastuzumab, la dosis recomendada de docetaxel es 100 mg/m2 cada 3 semanas, con trastuzumab administrado semanalmente. Para la determinación de la dosis y administración de trastuzumab, ver información para prescribir del producto. Como tratamiento de segunda linea, la dosis recomendada de docetaxel es 100 mg/m2 en monoterapia. Cuando se usa en combinacion con capecitabina, la dosis de TaxotereR es de 75 mg/m2 cada 3 semanas, asociado a capecitabina 1250 mg/m2 2 veces al dia por 2 semanas seguido de una semana de descanso, administrado oralmente (dentro de los 30 minutos después de las comidas). Para cálculo de dosis de capecitabina segun la superficie corporal, ver informacion para prescribir del producto. Cáncer de pulmón de células no pequeńas: en pacientes que no han sido tratados previamente con quimioterapia para cáncer de pulmón de células no pequeńas, el régimen de dosis recomendado es de 75 mg/m2, seguido inmediatamente por cisplatino 75 mg/m2 en 30-60 minutos o carboplatino (AUC 6 mg/ml x min.) en 30-60 minutos. Para el tratamiento después de haber fracasado a quimioterapias previas basadas en platinos, la dosis recomendada es 75 mg/m2 como monoterapia. Cáncer de ovario: Como tratamiento de segunda linea, la dosis recomendada de docetaxel es de 100 mg/m2 en monoterapia. Cáncer de cabeza y cuello: la dosis recomendada de docetaxel, combinado con cisplatino y 5-fluoruracilo, es de 75 mg/m2 administrada como infusión endovenosa de 1 hora, cada 3 semanas. Se recomienda la administración de una fluorquinolona oral, o antibióticos equivalentes en forma I.V., durante 10 días comenzando el día 5 de cada ciclo, para reducir infecciones/neutropenia febril. Quimioterapia de inducción seguida de radioterapia (TAX 323): Para el tratamiento de inducción del carcinoma de cabeza y cuello de células escamosas inoperable localmente avanzado, la dosis recomendada de Taxotere es 75 mg/m2 en infusión de 1 hora seguido de cisplatino 75 mg/m2 por 1 hora, seguido de 5-fluoruracilo como infusión continua de 750 mg/m2 por 5 dias. Este regimen se administra cada 3 semanas por 4 ciclos. A continuacion de la quimioterapia los pacientes deberian recibir radioterapia. Quimioterapia de inducción seguida de quimioradioterapia (TAX 324): Para el tratamiento de inducción de pacientes con carcinoma de cabeza y cuello de células escamosas inoperable localmente avanzado (no resecable, baja cura quirúrgica o con preservación del órgano), la dosis recomendada de Taxotere es 75 mg/m2 en infusión de 1 hora el día 1, seguido de cisplatino 100 mg/m2 administrado como infusión de 30 minutos a 3 horas, seguido de 5-fluoruracilo como infusión continua de 1000 mg/m2 desde el día 1 al 4. Este regimen se administra cada 3 semanas por 3 ciclos. A continuación de la quimioterapia los pacientes deberían recibir quimioradioterapia. Para modificación de la dosis de cisplatino y 5-fluoruracilo, ver su información para prescribir. Cáncer de próstata: la dosis recomendada de Taxotere® es 75 mg/m2 cada 3 semanas. Asociado a prednisona o prednisolona 5 mg 2 veces por día, vía oral, en forma continua. Adenocarcinoma gástrico: la dosis recomendada de Taxotere® es 75 mg/m2 como infusión endovenosa de 1 hora, seguido por cisplatino 75 mg/m2 como infusión de 1 a 3 horas (ambos sólo el día 1), seguido por 750 mg/m2 por día de 5-fluoruracilo administrado como infusión continua por 5 días, comenzando al término de la infusión de cisplatino. El tratamiento se repite cada 3 semanas. Los pacientes deben recibir premedicación con antieméticos e hidratación apropiada para la administración del cisplatino. Se puede usar G-CSF profiláctico para disminuir el riesgo de toxicidad hematológica. Ajustes de dosis durante el tratamiento: Generales: docetaxel debe ser administrado cuando el recuento de neutrófilos es ł 1.500 células/mm3. Los pacientes que han experimentado neutropenia febril, con recuento de neutrófilos < 500 células/mm3 por más de 1 semana, reacciones cutáneas graves o acumulativas, o neuropatía periférica aguda durante la terapia con docetaxel, la dosis de docetaxel debe ser reducida de 100 mg/m2 a 75 mg/m2 y/o de 75 a 60 mg/m2 . Si el paciente continúa experimentando estas reacciones a 60 mg/m2, el tratamiento debe ser suspendido. Terapia combinada con Taxotere® en cáncer de mama: pacientes que reciben terapia de adyuvancia para cáncer de mama y que presenten neutropenia febril deben recibir G-CSF en todos los ciclos siguientes. Los pacientes que persistan con esta reacción, deben continuar con G-CSF y reducir Taxotere® a 60 mg/m2. Si no se usa G-CSF, la dosis de Taxotere® debe reducirse desde 75 a 60 mg/m2. Pacientes que presenten estomatitis grado 3 ó 4 deberían disminuir la dosis a 60 mg/m2. Pacientes que reciben AC-TH o TCH como terapia de adyuvancia para cáncer de mama operable cuyos tumores sobreexpresen HER 2 y que presenten un episodio de neutropenia febril o infección deben recibir G-CSF profiláctico en todos los ciclos siguientes. Para un segundo episodio de neutropenia febril o infección, los pacientes deben continuar recibiendo G-CSF profiláctico, y reducir Taxotere desde 100 mg/m2 a 75 mg/m2 (en el régimen AC-TH); Taxotere sera reducido desde 75 mg/m2 a 60 mg/m2 (en el régimen TCH). Sin embargo, en la práctica clínica la neutropenia puede ocurrir en el ciclo 1. Por lo tanto, GCSF debe ser usado en consideración con el riesgo del paciente de hacer neutropenia y las recomendaciones actuales. Dependiendo del regimen de tratamiento, los pacientes que presenten estomatitis grado 3 ó 4, deberian reducir su dosis desde 100 mg/m2 a 75 mg/m2 (en el régimen AC-TH) o desde 75 mg/m2 a 60 mg/m2 (en el regimen TCH). Cuando se usa en combinación con capecitabina (para modificaciones de dosis de capecitabina, ver su folleto de información). Para los pacientes que desarrollan toxicidad Grado 2 por primera vez y que ésta persiste hasta el momento de la siguiente aplicación de Taxotere®/capecitabina, retrase el ciclo hasta que se resuelva a Grado 0-1, y reinicie con el 100% de la dosis original. Para los pacientes que desarrollan una segunda aparición de toxicidad Grado 2, o la primera de Grado 3, en cualquier momento del ciclo, retrase el tratamiento hasta que se resuelva a Grado 0 - 1, en ese momento reinicie el tratamiento con Taxotere® 55 mg/m2. Para la aparición de cualquier toxicidad subsecuente, o cualquier toxicidad grado 4, discontinúe la dosis de Taxotere®. Para modificaciones de dosis debida a toxicidad hepática, ver Precauciones. Terapia combinada con Taxotere® en cáncer de pulmón de células no pequeńas: para los pacientes que han sido tratados inicialmente con Taxotere® 75 mg/m2 en combinación con cisplatino o carboplatino, y en que han llegado a un recuento de plaquetas de < 25000/mm3 (con cisplatino) y de 75000/mm3 (con carboplatino), o en pacientes que presentan neutropenia febril, toxicidad no-hematológica severa, debe reducirse la dosis de Taxotere® en los ciclos siguientes a 65 mg/m2 (para modificaciones de dosis de cisplatino, ver su folleto de información). Terapia combinada con Taxotere® en cáncer gástrico: pacientes tratados con Taxotere® en combinación con cisplatino y 5-fluoruracilo deben recibir antieméticos e hidratación adecuada, de acuerdo con las directrices institucionales. A fin de mitigar el riesgo de neutropenia complicada se debería administrar G-CSF. Si a pesar del uso de G-CSF se presenta un episodio de neutropenia febril, neutropenia prolongada o infección neutropénica, se debe reducir la dosis de Taxotere® de 75 a 60 mg/m2. Si ocurren episodios subsecuentes de neutropenia complicada se debe reducir la dosis de Taxotere® de 60 a 45 mg/m2. En caso de trombocitopenia grado 4, la dosis de Taxotere® debe reducirse de 75 a 60 mg/m2. Los pacientes no deben ser retratados con ciclos subsecuentes de Taxotere® hasta recuperación del recuento de neutrófilos > 1500 células/mm3 y de plaquetas > 100000 células/mm3. Discontinuar el tratamiento si estas toxicidades persisten. Recomendaciones de modificación de dosis por toxicidad gastrointestinal en pacientes tratados con Taxotere en combinación con cisplatino y 5fluoruracilo: Toxicidad: Ajuste de dosis: Diarrea grado 3: Primer episodio: reducir la dosis de 5FU en 20% Segundo episodio: luego reducir dosis de Taxotere en 20%. Diarrea grado 4: Primer episodio: reducir dosis de Taxotere y 5FU en 20%. Segundo episodio: discontinuar tratamiento. Estomatitis grado 3: Primer episodio: reducir la dosis de 5FU en 20% Segundo episodio: suspender solamente 5FU, en todos los ciclos siguientes. Tercer episodio: reducir dosis de Taxotere en 20%. Estomatitis grado 4: Primer episodio: suspender solamente 5FU, en todos los ciclos siguientes. Segundo episodio: reducir dosis de Taxotere en 20%. Para modificaciones de dosis de cisplatino y fluoruracilo, ver su folleto de información. Poblaciones especiales: Ancianos: Sobre la base de un análisis farmacocinético de la población, no existen instrucciones especiales para su uso en ancianos. Para reducción de dosis de capecitabina cuando se combina con docetaxel, ver su folleto de información. Nińos: no se han establecido la seguridad y la eficacia de docetaxel en nińos. Pacientes con insuficiencia hepática: basándose en información farmacocinética con docetaxel a 100 mg/m2, como agente único, para los pacientes que tienen un incremento en los valores de transaminasas (ALT y/o AST) de más de 1.5 veces el límite superior normal e incrementos en fosfatasa alcalina superiores a 2.5 veces el límite superior normal, la dosis recomendada de docetaxel es de 75 mg/m2. Para aquellos pacientes con bilirrubina sérica > del límite superior normal y/o ALT y AST > 3.5 veces el límite superior normal, asociado con fosfatasa alcalina > 6 veces el límite superior normal, no se debe recomendar una reducción de dosis y, salvo una indicación precisa, no se debe usar docetaxel. No hay información disponible sobre pacientes con insuficiencia hepática tratadas con docetaxel en combinación.
|
|
Efectos Colaterales:
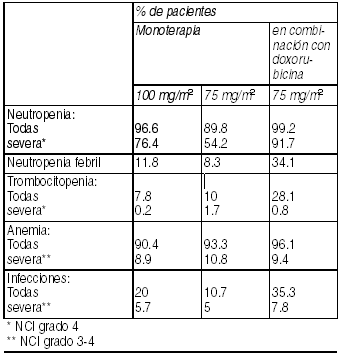
Las reacciones adversas consideradas como posible o probablemente relacionadas con la administración de docetaxel se han obtenido en pacientes tratados con docetaxel en monoterapia o en terapia combinada. Estos pacientes tenían prueba de función hepática basal normal. Estas reacciones fueron descritas utilizando Criterios Comunes de Toxicidad del NCI (Instituto Nacional del Cáncer) y términos COSTART (Símbolos de codificación para un diccionario de términos de reacciones adversas). Hematológicos: Se ha informado supresión de la médula ósea y otras reacciones hematológicas adversas que incluyen: Neutropenia (en pacientes que no recibieron GCSF), (96.6%). la neutropenia fue la reacción adversa más frecuente, reversible y no acumulativa La mediana de los días hasta el nadir fue de 7 días y la mediana de la duración de la neutropenia severa (< 500 células/mm3) (76.4%) fue de 7 días. Neutropenia febril (11.8%) e infección severa (4.6%) asociado con recuento de neutrófilos <500 células/mm3. Episodios infecciosos (20%) 5.7% severos incluyendo sepsis y neumonía fatal en 1.7%. Trombocitopenia <100.000/ mm3 (7.8%), (0.2% severa). Episodios de sangrado (2.4%) rara vez asociados con trombocitopenia grave (< 50000/mm3) y anemia (<11 g/dl: 90.4%), (8.9% severa (< 8 g/dl).
Ver Tabla
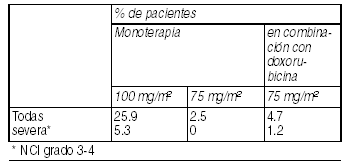
Se ha informado coagulación intravascular diseminada (CID), frecuentemente asociada con sepsis, o falla multiorgánica. Reacciones de hipersensibilidad: Las reacciones de hipersensibilidad generalmente han ocurrido a los pocos minutos del inicio de la infusión de docetaxel y, generalmente, fueron leves a moderadas (25.9%). Los síntomas informados más frecuentemente fueron rubor, sarpullido con o sin prurito, opresión torácica, dolor de espalda, disnea y fiebre inducida por droga o escalofríos. Las reacciones severas (5.3%), se resolvieron después de discontinuar la infusión y administrar terapia apropiada.
Ver Tabla
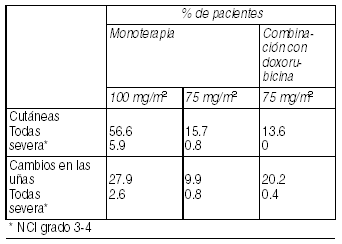
Raramente se informaron casos de shock anafiláctico. Muy rara vez estos casos tuvieron resultado fatal en pacientes que recibieron premedicación. Reacciones cutáneas: Se han observado reacciones cutáneas reversibles (56.6%), generalmente leves a moderadas. Las reacciones se caracterizaron por sarpullido, incluyendo erupciones localizadas principalmente en los pies, manos (incluyendo síndrome de pies y manos severo) pero también en los brazos, cara y tórax y asociada frecuentemente con prurito. Las erupciones generalmente se presentaron dentro de la semana de la infusión de docetaxel. Con menos frecuencia (5.9%), se informó sobre síntomas agudos tales como erupciones seguidas de descamación que raramente, necesitaron interrumpir o discontinuar el tratamiento con docetaxel. Las alteraciones de las uńas (27.9%)se caracterizan por hipo o hiperpigmentación, dolor y onicolisis. En casos aislados se informó sobre lupus eritematoso cutáneo y erupción bulosa, como eritema multiforme, síndrome de Stevens-Johnson, necrolisis tóxica epidérmica. En algunos casos, múltiples factores, tal como infecciones concomitantes, medicamentos concomitantes y enfermedad de base pueden haber contribuido al desarrollo de estos efectos.
Ver Tabla
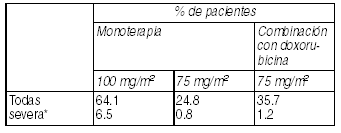
Retención de líquido: La retención de líquidos se ha observado en 64.1% (6.5% severo) de los pacientes que recibieron premedicación por 3 días. Se ha observado reacciones como edema periférico y, menos frecuentemente, derrame pleural, derrame pericárdico, ascitis y aumento de peso. El edema periférico, generalmente, comienza en las extremidades inferiores y puede llegar a ser generalizado, con un aumento de peso de 3 kg o más. La retención de fluidos es acumulativa en incidencia y en gravedad.
Ver Tabla
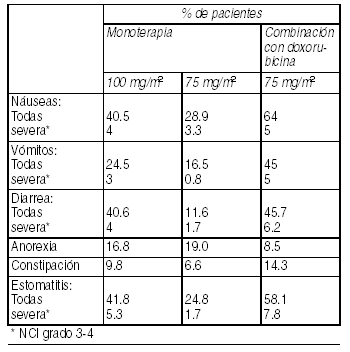
En pacientes tratados con Taxotere como agente único, a 100 mg/m2, la mediana de la dosis acumulada para interrupción del tratamiento fue más de 1000 mg/m2 y la mediana del tiempo en que la retención de fluidos revirtió fue de 16.4 semanas (rango de 0 a 42 semanas). El inicio de la retención moderada y severa se retrasa en los pacientes con premedicación (mediana de la dosis acumulativa: 818.9 mg/m2) en pacientes con premedicación, comparado con los pacientes sin premedicación (mediana de la dosis acumulativa: 489.7 mg/m2); sin embargo, se ha comunicado en algunos pacientes durante los primeros ciclos de terapia. La retención de líquidos no ha sido acompańada por episodios agudos de oliguria o hipotensión. En ocasiones aisladas se ha informado deshidratación y edema pulmonar. Trastornos gastrointestinales: Se han informado los siguientes eventos gastrointestinales: Náuseas (40.5% con 4% severo), vómitos (24.5 con 3% severo); diarrea (40.6 con 4% severo); dolor abdominal (7.3 con 1% severo), anorexia (16.8), constipación (9.8% con 0.2% severo), estomatitis (41.8 con 5.3% severo); esofagitis (1% con 0.4% severo), trastorno del gusto (10.15 con 0.07% severo), sangrado gastrointestinal (1.4% con 0.3% severo). Se han informado casos aislados de deshidratación a raíz de eventos gastrointestinales, perforación gastrointestinal, colitis isquémica, colitis y enterocolitis neutropénica. Se han informado casos aislados de obstrucción del ileon e intestinal.
Ver Tabla
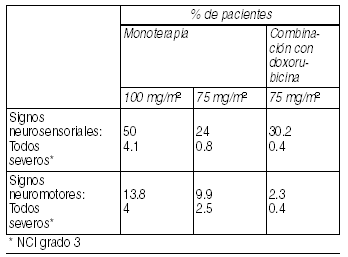
Reacciones neurológicas: Se han presentado signos y/o síntomas neurosensoriales leves a moderados en un 50% de los pacientes. Síntomas neurosensoriales graves (parestesia, disestesias ó dolor con sensación de quemazón) fueron observados en 4.1% de los pacientes con cáncer de mama metastático y llevaron a discontinuar el tratamiento en 2%. Los eventos neuromotores (13.8% con 4% severos) se caracterizan, principalmente, por debilidad. Cuando aparece esta sintomatología se requiere ajustar la dosis. Si los síntomas persisten el tratamiento debe discontinuarse. Aquellos pacientes que presentaron neurotoxicidad durante estudios clínicos y para los que hay información de seguimiento hasta resolución completa del evento, presentaron remisión espontánea de los síntomas con un promedio de 81 días desde el inicio (rango: 0 a 741 días). Con la administración de docetaxel se han observado casos aislados de convulsiones o pérdida transitoria del conocimiento. Estas reacciones aparecen algunas veces durante la infusión de la droga.
Ver Tabla
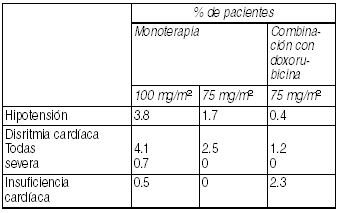
Trastornos cardiovasculares: Los eventos cardiovasculares consisten en: Hipotensión, disritmia, hipertensión, insuficiencia cardíaca. Se han informado casos aislados de eventos tromboembólicos venosos, como así también de infarto del miocardio.
Ver Tabla
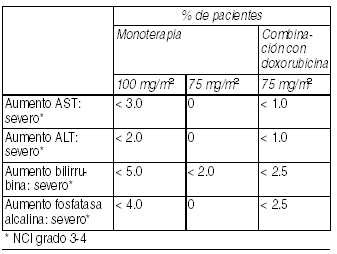
Trastornos hepáticos: En pacientes tratados con 100 mg/m2 en monoterapia, en menos del 5% de las pacientes se observaron incrementos en los niveles séricos de GOT, GPT, bilirrubina y fosfatasa alcalinas mayores a 2.5 veces el límite superior del rango normal. Se ha informado de casos muy raros de hepatitis, algunas veces fatal principalmente en pacientes con trastornos hepáticos preexistentes.
Ver Tabla
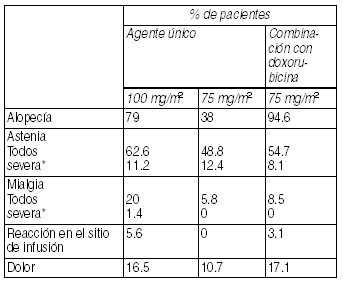
Trastornos del oído y laberinto: Se ha informado de raros casos de ototoxicidad, trastornos auditivos y/o pérdida de la audición, incluyendo casos asociados con otras drogas ototóxicas. Otros: Alopecia (79% con 0.5% severo); astenia (62.2% con 11.2% severo); artralgias (8.6%); mialgias (20%); disnea (16.1% con 2.7 severa). Dolor generalizado o localizado (16.3% con 0.8% severo), incluyendo dolor torácico (4.5% con 0.4% severo) con o sin compromiso cardíaco o respiratorio. Rara vez se han informado casos de lagrimeo con o sin conjuntivitis, así como muy raros casos de obstrucción del conducto lagrimal con el consecuente lagrimeo excesivo, principalmente en pacientes que están recibiendo concomitantemente otros agentes antitumorales. Se han informado casos muy aislados de trastornos visuales transitorios (relampagueos, luces titilantes, escotomas) que se presentan generalmente durante la infusión de la droga y asociados a reacciones de hipersensibilidad. Estos se revirtieron al discontinuar la infusión. Las reacciones en el sitio de infusión fueron generalmente leves, consistentes en hiperpigmentación, inflamación, enrojecimiento o sequedad de la piel, flebitis o extravasación y tumefacción de la vena. Aisladamente, se han informado casos de síndrome de insuficiencia respiratoria aguda, neumonía intersticial, fibrosis pulmonar y fenómeno de recuerdo de radiación. En pacientes que reciben radioterapia concomitante se han reportado raros casos de neumonitis por radiación.
Ver Tabla
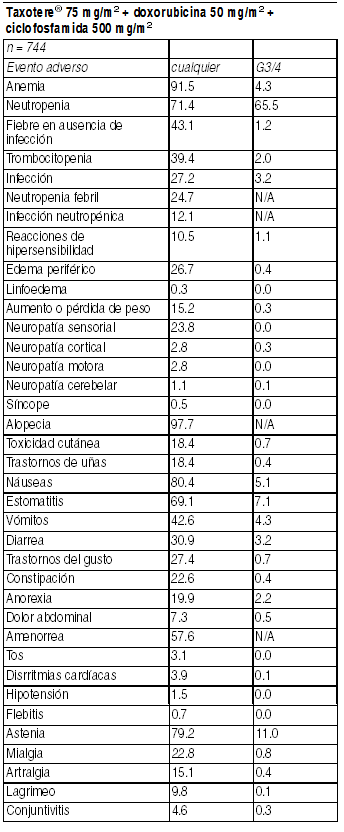
En general, el patrón de eventos adversos observado en pacientes tratados con Taxotere en combinación con doxorubicina es similar al de aquellos tratados con monoterapia. Terapia combinada de Taxotere en tratamiento adyuvante para cáncer de mama: La siguiente tabla presenta eventos clínicamente importantes relacionados al tratamiento, observados en 744 pacientes que recibieron Taxotere 75 mg/m2 cada 3 semanas con la combinación doxorubicina y ciclofosfamida. Eventos adversos clínicamente importantes en pacientes que recibieron Taxotere en combinación doxorubicina y ciclofosfamida (TAX316):
Ver Tabla
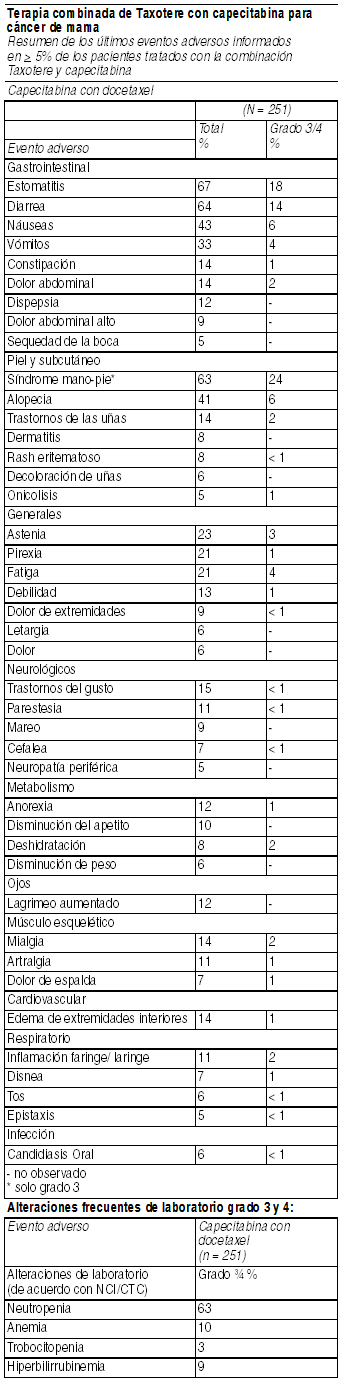
De los 744 pacientes tratados con TAC, 33.1% presentó eventos adversos serios procedentes del tratamiento. Reducciones de dosis debido a toxicidad hematológica ocurrieron en 1% de los ciclos. 6% de los pacientes discontinuaron tratamiento debido a la ocurrencia de eventos adversos; fiebre en ausencia de infección y alergia fueron las razones más comunes de abandono. Dos pacientes murieron dentro de los últimos 30 días de tratamiento, 1 muerte fue considerada relacionada con la droga de estudio. Fiebre e infección: Fiebre en ausencia de infección se observó en 43.1% (G3/4: 1.3%) de los pacientes e infección fue observada en 27.2% (G3/4: 3.9%) de los pacientes. No hubo muertes por sepsis. Eventos gastrointestinales: Además de los eventos gastrointestinales de la tabla anterior, 4 pacientes reportaron colitis/ enteritis/ perforación del intestino grueso. Dos de estos pacientes requirieron discontinuar el tratamiento; no se informaron muertes por esta causa. Eventos cardiovasculares: Se informaron los siguientes eventos cardiovasculares: arritmias, todos los grados (3.9%), hipotensión todos los grados (1.5%), e insuficiencia cardíaca congestiva (2.3% en un seguimieto medio de 70 meses). Un paciente murió por insuficiencia cardíaca. Leucemia mieloide aguda (LMA)a/ síndrome mielo displástico. En un tiempo promedio de seguimiento de 83 meses, 3 de 744 (0.4%) pacientes que recibieron Taxotere, doxorubicina, y ciclofosfamida pacientes fueron diagnosticados con LMA y en 1 de 736 (0.1%) pacientes que recibieron fluoruracilo, doxorubicina y ciclofosfamida. Experiencia post-comercialización: Se han reportado casos muy raros de LMA y de síndrome mielodisplástico en asociación con docetaxel usado en combinación con otros agentes quimioterápicos y/o radioterapia. Otras reacciones persistentes: Los siguientes eventos fueron observados en el curso del período de seguimiento de 55 meses: alopecia (22/687), amenorrea (133/233), edema neurosensorial (9/73) y periférico (18/112). Terapia combinada de Taxotere con capecitabina para cáncer de mama. Resumen de los últimos eventos adversos informados en 5% de los pacientes tratados con la combinación Taxotere y capecitabina:
Ver Tabla
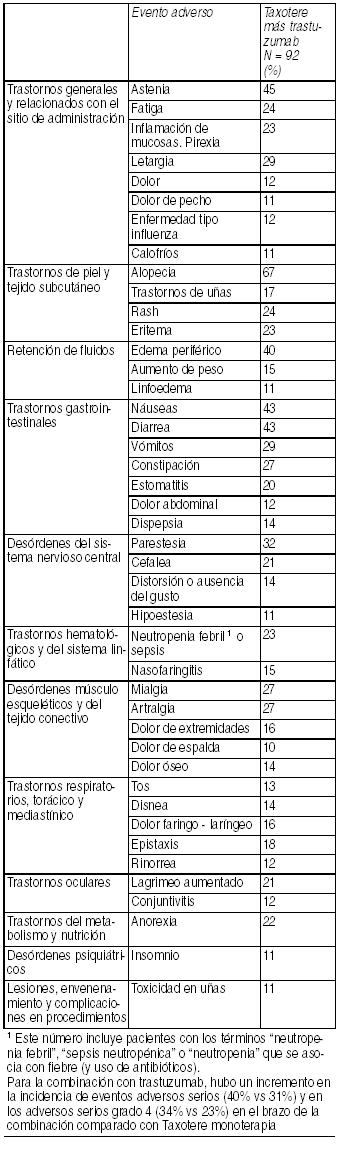
Terapia combinada de Taxotere con trastuzumab para cáncer de mama: La siguiente tabla muestra los eventos adversos(todos los grados), que fueron reportados en ł 10% de los pacientes con Taxotere y trastuzumab para cáncer de mama metastásico:
Ver Tabla
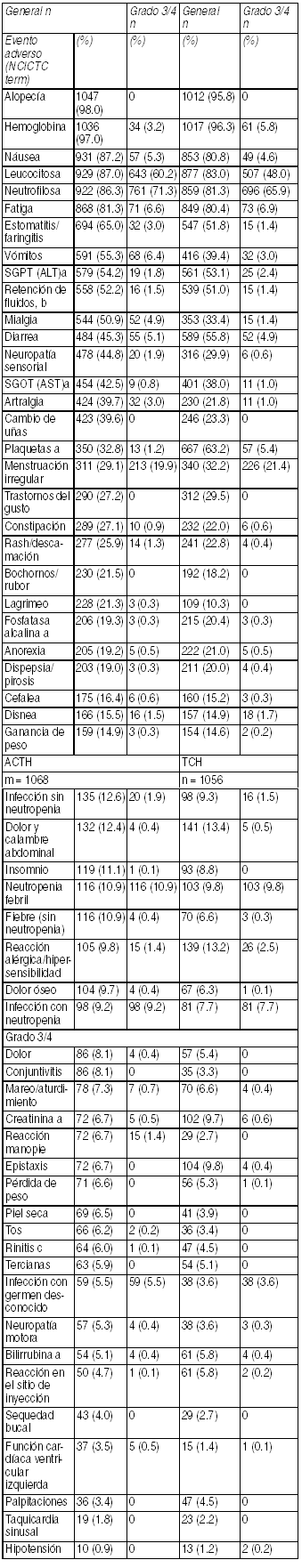
Toxicidad cardíaca: Fue reportada insuficiencia cardíaca sintomática en 2.2% de los pacientes que recibieron Taxotere más trastuzumab comparado con 0% de pacientes que recibieron Taxotere solo. En el brazo Taxotere más trastuzumab, 64% había recibido antraciclinas cono terapia de adyuvancia, comparado con 55% en el brazo sólo con Taxotere. Toxicidad hematológica: Neutropenia grado 3/4 fue reportada en 32% de los pacientes con Taxotere más trastuzumab. Terapia combinada de Taxotere® para el tratamiento adyuvante de pacientes con cáncer de mama operable cuyos tumores sobreexpresen HER 2 que recibieron ACTH o TCH. Eventos adversos relacionados al tratamiento en estudio, con ocurrencia en cualquier momento durante ele estudio: población de seguridad (incidencia ł 5% para eventos no cardíacos; incidencia ł 1% para eventos cardíacos) ACTH TCH n=1068 n=1056.
Ver Tabla
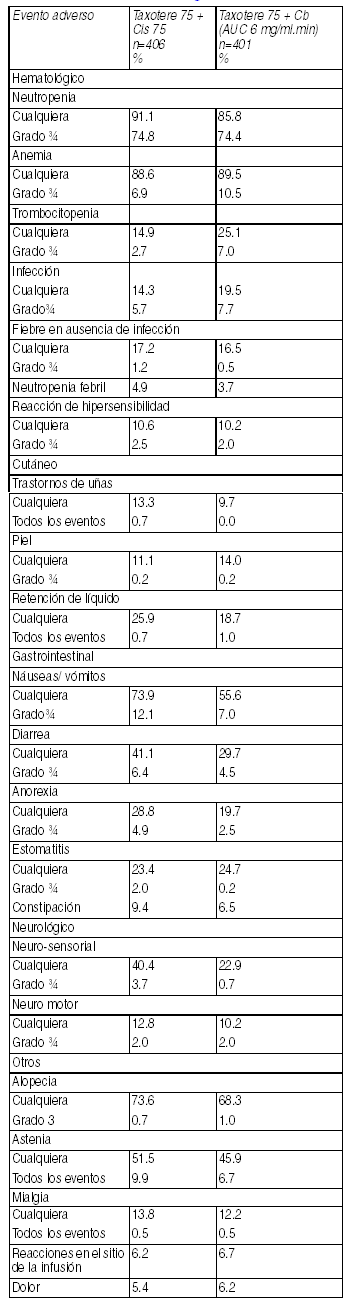
ACTH = doxorubicina y ciclofosfamida, seguido de Taxotere en combinación con trastuzumab. TCH = Taxotere en combinación con trastuzumab y carboplatino. a independiente de causalidad. b Los eventos adversos de retención de fluidos se definen como "sólo edema ", o "sólo ganancia de peso", o "sólo edema pulmonar", o "edema y ganancia de peso", o "edema y edema pulmonar", o "edema + ganancia de peso + edema pulmonar". "retención de fluidos " corresponde al término NCICTC "edema"; c término COSTART. Terapia combinada de Taxotere en pacientes con cáncer de pulmón de células no-pequeńas: Eventos adversos clínicamente importantes relacionados al tratamiento en pacientes con cáncer de pulmón de células no pequeńas recibiendo Taxotere en combinación con cisplatino(Cis) o Carboplatino(Cb). Terapia combinada de Taxotere en pacientes con cáncer de próstata:
Ver Tabla
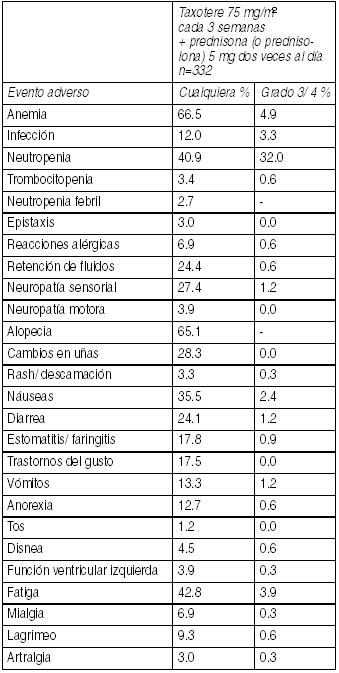
La siguiente información está basada en la experiencia con 332 pacientes, que fueron tratados con 75 mg/m2 Taxotere cada 3 semanas en combinación con 5 mg de prednisona o prednisolona oral 2 veces al día. Eventos adversos clínicamente importantes relacionados al tratamiento en pacientes con cáncer de próstata que recibieron Taxotere en combinación con prednisona o prednisolona (TAX 327):
Ver Tabla
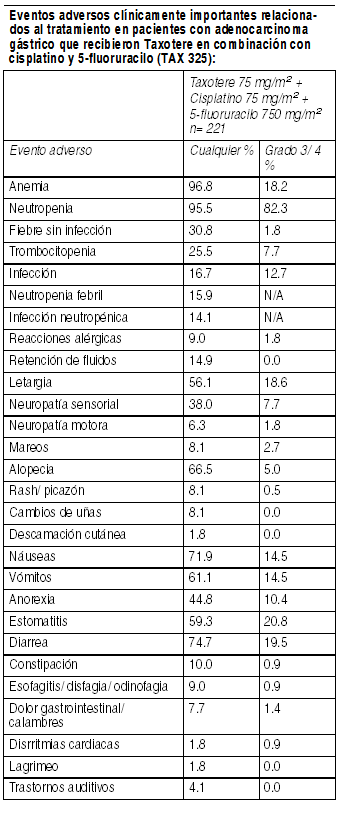
Terapia combinada de Taxotere en pacientes con adenocarcinoma gástrico: La siguiente información está basada en la experiencia con 221 pacientes con adenocarcinoma gástrico avanzado y sin antecedentes de quimioterapia previa para enfermedad avanzada, que fueron tratados con 75 mg/m2 Taxotere en combinación con cisplatino y 5fluoruracilo.
Ver Tabla
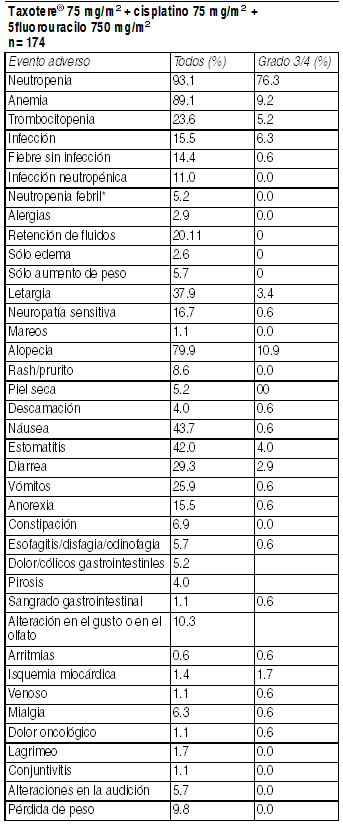
La neutropenia febril y/o Infección neutropénica ocurrió en 28.6% de los pacientes independientemente del uso de GCSF, el GCSF fue usado para profilaxis secundaria en sólo 18.6% de los pacientes (10% de los ciclos) en el brazo TCF. La neutropenia febril y/o Infección neutropénica ocurrió en tasas bajas, 12.2% cuando los pacientes recibieron GCSF, y en 26.9% sin profilaxis con GCSF. Tratamiento combinado con Taxotere® en cáncer de cabeza y cuello: La siguiente tabla resume los datos de seguridad obtenidos en 174 pacientes con cáncer de cabeza y cuello de células escamosas localmente avanzado e irresecable, quienes fueron tratados con Taxotere® 75 mg/m2 en combinación con cisplatino y 5fluorouracilo. Eventos adversos clínicamente importantes relacionados al tratamiento en pacientes con cáncer de cabeza y cuello que recibieron Taxotere en combinación con cisplatino y 5fluoruracilo (TAX 323):
Ver Tabla
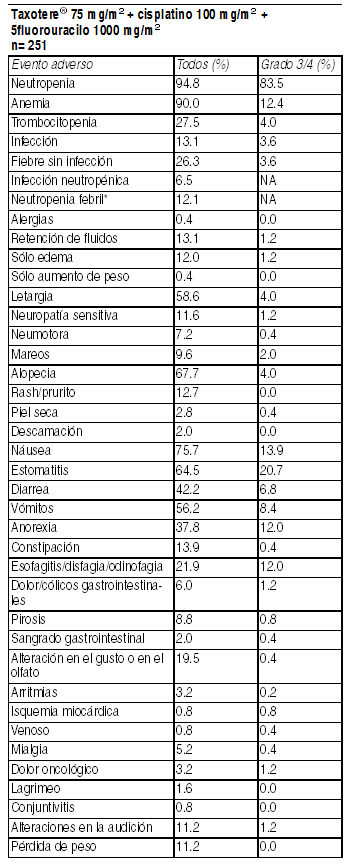
Neutropenia febril: grado ł 2 con fiebre concomitante con neutropenia grado 4 que requiera hospitalización y/o antibióticos I.V. Eventos adversos clínicamente importantes relacionados al tratamiento en pacientes con cáncer de cabeza y cuello que recibieron Taxotere en combinación con cisplatino y 5fluoruracilo (TAX 324). Neutropenia febril: grado ł 2 con fiebre concomitante con neutropenia grado 4 que requiera hospitalización y/o antibióticos I.V.
Ver Tabla
|
|
Contraindicaciones:
Taxotere está contraindicado en: Pacientes con historia de reacciones de hipersensibilidad severa a la droga o al polisorbato 80. Pacientes con recuento basal de neutrófilos <1.500 células/mm3. Embarazo. Lactancia. Pacientes con insuficiencia hepática grave. Las contraindicaciones de otros medicamentos son aplicables cuando se los combina con docetaxel.
|
|
Precauciones:
Precauciones especiales para su uso: La incidencia y la gravedad de la retención de líquidos, así como la gravedad de las reacciones de hipersensibilidad pueden reducirse por la premedicación con corticoesteroides orales, como dexametasona 16 mg diarios (ej. 8 mg 2 veces al día) durante 3 días, empezando 1 día antes de la administración de docetaxel, a no ser que haya contraindicaciones. Para el cáncer de próstata, la premedicación es dexametasona oral 8 mg, 12 horas, 3 horas y 1 hora antes de la infusión de docetaxel. Neutropenia: El nadir de neutrófilos ocurre en una mediana de 7 días, pero este intervalo puede acortarse en pacientes pretratados intensamente. En todos los pacientes tratados con docetaxel se debe realizar un monitoreo frecuente y completo del recuento sanguíneo. Los pacientes deben ser retratados con docetaxel sólo cuando los neutrófilos se recuperan a un nivel de ł 1.500 células/mm3. En pacientes tratados con Taxotere en combinación con cisplatino y 5fluoruracilo (TCF), la neutropenia febril y/o infección neutropénica se redujo cuando recibieron GCSF en forma profiláctica. Los pacientes tratados con TCF deberían recibir GCSF en forma profiláctica para mitigar el riesgo de neutropenia complicada (neutropenia febril, neutropenia prolongada o infección neutropénica). Los pacientes que reciban TCF deben ser estrechamente vigilados. Reacciones de hipersensibilidad: Los pacientes deben ser supervisados muy de cerca por reacciones de hipersensibilidad, especialmente durante las primeras y segundas infusiones. Las reacciones de hipersensibilidad se pueden presentar a los pocos minutos de iniciarse la infusión con docetaxel, por consiguiente es necesario tener disponible instalaciones para el tratamiento de hipotensión y broncoespasmo. Cuando ocurre reacciones graves, tales como hipotensión grave, broncoespasmo, o rash cutáneo / eritema generalizado se debe discontinuar inmediatamente el docetaxel y aplicar una terapia adecuada. Los pacientes que han desarrollado reacciones de hipersensibilidad grave no deben volver a tratarse con docetaxel. Reacciones cutáneas: Se ha observado eritema cutáneo localizado de las extremidades (palmas de las manos y plantas de los pies) con edema, seguido por descamación. Retención de líquidos. Se debe supervisar estrechamente a los pacientes con retención severa de líquidos tales como derrame pleural, derrame pericárdico y ascitis. Pacientes con insuficiencia hepática: En los pacientes tratados con docetaxel 100 mg/m2 como agente único, que presentan niveles de transaminasa sérica (ALT y/o AST) > 1.5 veces el límite superior normal, coincidente con niveles de fosfatasa alcalina sérica > 2.5 veces el límite superior normal, existe un mayor riesgo de desarrollar reacciones adversas graves, tales como muertes tóxicas incluyendo sepsis y hemorragia gastrointestinal, neutropenia febril, infecciones, trombocitopenia, estomatitis y astenia. Por consiguiente, la dosis recomendada de docetaxel en aquellos pacientes con pruebas de la función hepática elevadas es 75 mg/m2. La función hepática debe evaluarse antes de iniciar la terapia y antes de cada ciclo. No se recomienda reducción de dosis y docetaxel no debe ser utilizado, salvo indicación precisa, para pacientes con niveles de bilirrubina sérica > límite superior normal y/o ALT y AST >3.5 veces el límite superior normal coincidente con niveles de fosfatasa alcalina sérica > 6 veces el ULN. No hay información disponible en pacientes con insuficiencia hepática tratados con docetaxel en terapia combinada. Sistema nervioso: Se ha observado desarrollo de signos y/o síntomas neurosensoriales severos y requieren reducción de dosis. Toxicidad cardíaca: Se ha observado insuficiencia cardíaca, en pacientes que han recibido Taxotere® en combinación con trastuzumab, particularmente después de quimioterapia que incluyeron antraciclinas (doxorrubicina o epirrubicina). Esta puede ser moderada a severa y se ha asociado con muerte. Leucemia: En el tratamiento adjuvante de cáncer de mama, el riesgo de mielodisplasia tardía o leucemia mieloide requiere seguimiento hematológico. Ancianos: Un análisis de seguridad: los pacientes de 60 ańos o más tratados con terapia combinada de Taxotere® + capecitabina, mostraron una mayor incidencia de eventos adversos Grado 3 - 4 relacionados al tratamiento, eventos adversos serios relacionados y suspensión anticipada del tratamiento debido a eventos adversos, cuando se comparó con pacientes < 60 ańos. En los regímenes ACTH y TCH la proporción de pacientes ancianos fue 5.5% y 6.6%, respectivamente y es muy limitada para permitir sacar conclusiones sobre la ocurrencia de eventos adversos por edad (< 65 ańos versus ł 65 ańos). En un estudio de cáncer de próstata, de los 333 pacientes tratados con Taxotere® cada 3 semanas, 209 tenían 65 ańos o más y 68 eran mayores de 75 ańos. No se identificaron diferencias en eficacia entre pacientes ancianos y más jóvenes. La incidencia de la anemia, infecciones, alteraciones en las uńas, anorexia, pérdida de peso se presentó con una tasa de incidencia ł 10% más elevados en pacientes que tenían 65 ańos o más versus pacientes más jóvenes. Otras: Durante la terapia y, por lo menos por 3 meses después se debe adoptar métodos anticonceptivos adecuados. Reacciones gastrointestinales: Síntomas tempranos como dolor y sensibilidad abdominal, fiebre, diarrea, con o sin neutropenia pueden ser manifestaciones tempranas de toxicidad gastrointestinal grave y deben ser valoradas y tratadas de forma inmediata. Pacientes con 4 o más nódulos: La relación riesgo/beneficio para TAC en pacientes con 4 ó + nódulos no está completamente definida en el análisis intermedio. Embarazo: Se ha determinado que docetaxel es tanto embriotóxico como fetotóxico en conejos y ratas y que disminuye la fertilidad en ratas. Docetaxel puede causar dańos al feto cuando se lo administra a mujeres embarazadas. Por consiguiente, docetaxel no debe ser administrado a mujeres embarazadas. Se debe advertir a las mujeres en edad fértil que eviten quedar embarazadas si están recibiendo docetaxel y que deben informar inmediatamente al médico tratante si esto ocurre. Lactancia: Se desconoce si docetaxel se excreta en la leche humana. Debido al riesgo potencial de reacciones adversas en los lactantes, se debe discontinuar el amamantamiento durante la terapia de docetaxel.
|
|
Interacciones Medicamentosas:
Los estudios in vitro han demostrado que el metabolismo de docetaxel puede ser modificado por la administración concomitante de compuestos que inducen, inhiben o son metabolizados por el citocromo P4503A, tales como ciclosporina, terfenadina, ketoconazol, eritromicina y troleandomicina. Por lo tanto, es necesario tener precaución al tratar a pacientes con estas drogas como terapia concomitante, pues existe un potencial de una interacción significativa. La unión de docetaxel a proteínas es elevada (>95%). Si bien no se ha investigado formalmente la posible interacción in vivo de docetaxel con la medicación administrada concomitantemente, las interacciones in vitro con drogas que se unen fuertemente a proteínas, tales como eritromicina, difenhidramina, propranolol, propafenona, fenitoína, salicilato, sulfametoxazol y valproato sódico no afectaron la unión a proteína de docetaxel. Además, dexametasona no afectó la unión a proteína de docetaxel ni tuvo ninguna influencia sobre la unión de digitoxina.
|
|
Sobredosificación:
Hay muy pocos informes de sobredosis. En caso de sobredosis, se debe mantener al paciente en una unidad especializada donde se puedan monitorear las funciones vitales. No existe ningún antídoto conocido para el caso de sobredosis con docetaxel. Las complicaciones primarias que se prevén por sobredosis incluirían: supresión de médula ósea, neurotoxicidad periférica y mucositis. Luego de descubrir la sobredosificación, los pacientes deben recibir GCSF terapéutico tan pronto como sea posible. Según sea necesario, se deben adoptar medidas sintomáticas apropiadas.
|
|
Conservación:
Los viales deben ser guardado a una temperatura de entre 2şC y 25şC y protegidos de la luz brillante. Condiciones de conservación y almacenamiento: Frasco-ampolla sin abrir: entre (+2 y +25)şC. Mantener en el envase original como protección de la acción de luz intensa. El congelamiento no afecta adversamente al producto. Solución Premezcla (10 mg docetaxel/ml): se puede utilizar inmediatamente o almacenar durante un máximo de 8 horas, ya sea a temperatura ambiente o en refrigeración (entre +2°C y +8°C) para la preparación de la solución para infusión. La solución para infusión ya preparada (ya sea en solución 0.9% de cloruro de sodio o en solución 5% de dextrosa) deberá usarse dentro de las 4 horas (incluyendo la hora de administración I.V.).
|
|
Observaciones:
Vial de Taxotere 80 mg concentrado de solución para infusión: el vial de Taxotere 80 mg concentrado de solución para infusión es un frasco de vidrio transparente de 15 ml con una tapa dispensadora especial (flip-off) verde. Este vial contiene 2 ml de una solución de 40 mg/ml de docetaxel en polisorbato 80 (volumen de llenado: 94.4 mg/2.36 ml). Este volumen de llenado ha sido establecido durante el desarrollo de Taxotere para compensar la pérdida de líquido durante preparación de la premezcla debido a formación de espumas, adhesión a las paredes del vial y volumen inerte. Este sobrellenado garantiza que, luego de la dilución con el contenido total del diluyente que acompańa el vial de Taxotere, hay un volumen mínimo de premezcla extraíble de 2 ml conteniendo 10 mg/ml de docetaxel que corresponde a la cantidad del rotulado de 80 mg por vial. Vial de diluyente: el vial de diluyente es un frasco de vidrio transparente de 15 ml con una tapa dispensadora especial (flip-off) incolora. El vial de diluyente contiene 6 ml de solución al 13% p/p de etanol en agua para inyectables (volumen de llenado: 7.33 ml). El ańadido de todo el contenido del vial de diluyente al contenido del vial de Taxotere 80 mg concentrado de solución para infusión asegura una concentración de premezcla de 10 mg/ml de docetaxel. Vial de Taxotere 20 mg: el vial de Taxotere 20 mg es un frasco de vidrio transparente de 7 ml con una tapa dispensadora especial (flip-off) verde. Este vial contiene 0.5 ml de una solución de 40 mg/ml de docetaxel en polisorbato 80 (volumen de llenado: 24.4 mg/0.61 ml). Este volumen de llenado ha sido establecido durante el desarrollo de Taxotere para compensar la pérdida de líquido durante preparación de la premezcla debido a formación de espumas, adhesión a las paredes del vial y volumen inerte. Este sobrellenado garantiza que, luego de la dilución con el contenido total del diluyente que acompańa el vial de Taxotere, hay un volumen mínimo de premezcla extraíble de 2 ml conteniendo 10 mg/ml de docetaxel que corresponde a la cantidad del rotulado de 20 mg por vial. Vial de diluyente: el vial de diluyente para Taxotere 20 mg es un frasco de vidrio transparente de 7 ml con una tapa dispensadora especial (flip-off) incolora. El vial de diluyente contiene 1.5 ml de solución al 13% p/p de etanol en agua para inyectables (volumen de llenado 1.98 ml). El ańadido de todo el contenido del vial de diluyente al contenido del vial de Taxotere 20 mg asegura una concentración de premezcla de 10 mg/ml de docetaxel. Instrucciones para uso, manipulación y eliminación: Recomendaciones para la manipulación segura: Taxotere es un medicamento antineoplásico y, al igual que con otros compuestos potencialmente tóxicos, se debe ejercer precaución en su manipulación y en la preparación de las soluciones de Taxotere. Se recomienda el uso de guantes. Si se produce contacto de la piel con el concentrado de Taxotere, la solución premezcla o la solución para infusión, lavar inmediata y minuciosamente con agua y jabón. Si el concentrado de Taxotere, la solución premezcla o la solución para infusión entra en contacto con las membranas mucosas, lavar inmediata y minuciosamente con agua. Preparación de la administración I.V.: Preparación de la solución premezcla de Taxotere (10 mg docetaxel/ml): si los viales están guardados en un refrigerador, dejar que la cantidad necesaria de cajas permanezca a temperatura ambiente durante 5 minutos. Extraer asépticamente todo el contenido del vial de diluyente para Taxotere, utilizando una jeringa con una aguja incorporada, invirtiendo parcialmente el vial. Inyectar todo el contenido de la jeringa en el vial correspondiente a Taxotere. Quitar la jeringa y aguja y mezclar manualmente mediante repetidas inversiones durante, por lo menos, 45 segundos. No agitar. Permitir que el vial de premezcla repose durante 5 minutos a temperatura ambiente y luego controlar que la solución sea homogénea y transparente (la formación de espuma es normal aún después de 5 minutos debido a la presencia del polisorbato 80 en la formulación). La solución premezcla contiene 10 mg/ml de docetaxel y debe ser usada inmediatamente después de preparada. Sin embargo, se ha demostrado su estabilidad química y física de 8 horas cuando se la almacena entre 2şC y 8şC o a temperatura ambiente. Preparación de la solución para infusión: puede ser necesario más de un vial de solución premezcla para obtener la dosis requerida para el paciente. En base a la dosis en mg requerida por el paciente, retirar asépticamente el correspondiente volumen premezcla conteniendo 10 mg/ml de docetaxel de la cantidad adecuada de viales de solución premezcla utilizando jeringas graduadas con aguja incorporada. Por ejemplo, una dosis de 140 mg de docetaxel necesitaría 14 ml de solución premezcla de docetaxel. Inyectar el volumen de solución premezcla a una bolsa o frasco de infusión de 250 ml conteniendo 5% de solución de glucosa o 0.9% de solución de cloruro sódico. Si se necesita una dosis mayor de 200 mg de docetaxel, utilizar una bolsa de infusión de mayor volumen para que no se exceda una concentración de 0.74 mg/ml de docetaxel. Mezclar la bolsa o frasco de infusión por medio de un movimiento de rotación. La solución para infusión de Taxotere debe ser usada dentro de las 4 horas y debe ser administrada asépticamente como una infusión de 1 hora, a condiciones de temperatura ambiente y luz normal. Al igual que todos los productos parenterales, la solución Taxotere premezcla y solución para infusión deben ser inspeccionadas visualmente antes de su uso. Las soluciones que contienen un precipitado deben ser eliminadas. Eliminación: todos los materiales que han sido utilizados para dilución y administración deben ser eliminados conforme a procedimientos estándar.
|
|
Presentaciones:
Taxotere 20 mg: envase conteniendo 1 frasco-ampolla monodosis y 1 frasco-ampolla monodosis de 1.5 ml de diluyente. Ambos se expanden en un mismo blíster, dentro de un estuche. Taxotere 80 mg: envase conteniendo 1 frasco-ampolla monodosis y 1 frasco-ampolla monodosis de 6 ml de diluyente, ambos se expanden en un mismo blíster, dentro de un estuche.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}