 |
Composición:
Cada comprimido recubierto contiene: Abacavir (como sulfato de abacavir) 300 mg; Lamivudina 150 mg y Zidovudina 300 mg. Los comprimidos recubiertos con forma de cápsula son de color azul verdoso y grabados en una cara con GX LL1. Excipientes: Núcleo: Celulosa Microcristalina; Almidón Glicolato Sódico; Estearato de Magnesio. Recubrimiento: Opadry Verde 03B11434 que contiene: Hipromelosa; Dióxido de Titanio; Macrogol; Colorante FD&C Azul Nş2; Laca Alumínica; Oxido de Hierro Amarillo.
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: Análogo de nucleósido. Código ATC: J05A R04.
|
|
Indicaciones:
Tricivir® comprimidos es una combinación de tres análogos de nucleósidos (abacavir, lamivudina y zidovudina). Está indicado para el tratamiento antirretroviral de la infección por el Virus de Inmunodeficiencia Humano (VIH) en adultos y adolescentes mayores de 12 ańos infectados, y que tengan un peso superior a los 40 kg.
|
|
Propiedades:
Mecanismo de acción y resistencia: Lamivudina, zidovudina y abacavir son nucleósidos análogos inhibidores de la transcriptasa inversa y son potentes inhibidores selectivos de HIV-1 y HIV-2. Los tres medicamentos se metabolizan secuencialmente por medio de cinasas intracelulares para el 5'-trifosfato (TP) respectivo. Lamivudina-TP, abacavir-TP y zidovudina-TP son sustratos para la transcriptasa inversa del VIH e inhibidores competitivos de la misma. Sin embargo, su principal actividad antiviral es por medio de la incorporación de la forma monofosfato en la cadena de ADN viral, lo que conduce a la terminación de la cadena. Los trifosfatos de lamivudina, abacavir y zidovudina muestran una afinidad significativamente menor por las ADN polimerasas de la célula hospedadora. Lamivudina ha mostrado un elevado sinergismo con zidovudina, inhibiendo la replicación del VIH en cultivos celulares. Abacavir muestra sinergia in vitro en combinación con zidovudina y ha demostrado ser aditivo en combinación con lamivudina. Efectos farmacodinámicos: Se han seleccionado in vitro aislados de VIH-1 resistentes a abacavir y se han relacionado con cambios genotípicos específicos en la región del codón (codones M184V, K65R, L74V y Y115F) de la transcriptasa inversa (TI). La resistencia viral a abacavir se desarrolla relativamente despacio in vitro e in vivo, precisando múltiples mutaciones para alcanzar un incremento de ocho veces en la CI50 sobre el virus de tipo silvestre, el cual puede ser un nivel clínicamente importante. Los aislados resistentes a abacavir pueden mostrar también una sensibilidad reducida a lamivudina, zalcitabina y/o didanosina, pero permanecen sensibles a zidovudina y estavudina. Una falla en el tratamiento después de una terapia de combinación inicial con abacavir, lamivudina y zidovudina, se asocia principalmente con el M184V solo, manteniendo así muchas opciones de tratamiento para un régimen de segunda línea. No es probable la aparición de resistencias cruzadas entre abacavir, lamivudina o zidovudina e inhibidores de la proteasa o inhibidores de la transcriptasa inversa no análogos de nucleósidos. Se ha demostrado una sensibilidad reducida a abacavir en aislados clínicos de pacientes con replicación viral no controlada tratados previamente con otros inhibidores análogos de nucleósidos y resistentes a ellos. Propiedades farmacocinéticas: Absorción: Lamivudina, abacavir y zidovudina se absorben bien y rápidamente en el tracto gastrointestinal tras su administración oral. La biodisponibilidad absoluta de lamivudina, abacavir y zidovudina administrados por vía oral en adultos es de, aproximadamente, 80-85%, 83 % y del 60-70% respectivamente. En un estudio farmacocinético realizado en pacientes infectados por el VIH-1, los parámetros farmacocinéticos en el estado de equilibrio de abacavir, lamivudina y zidovudina fueron similares tanto cuando se administró Tricivir® comprimidos solo, como cuando se administró la combinación de Combivir y Ziagen®. Los parámetros en estado de equilibrio también fueron similares a los valores obtenidos en el estudio de bioequivalencia de Tricivir® comprimidos en voluntarios sanos. Un estudio de bioequivalencia comparó Combivir con comprimidos de 150 mg de lamivudina y de 300 mg de zidovudina administrados al mismo tiempo. También se estudió el efecto de los alimentos sobre la tasa y el grado de absorción. Combivir demostró ser bioequivalente a lamivudina 150 mg y zidovudina 300 mg, administrados en comprimidos por separado, al ser administrados a individuos en ayunas. En un estudio de bioequivalencia se comparó Tricivir® comprimidos con lamivudina150 mg, zidovudina 300 mg y abacavir 300 mg administrados al mismo tiempo. También se estudió el efecto de los alimentos sobre la tasa y el grado de absorción. Tricivir® comprimidos demostró ser bioequivalente a lamivudina 150 mg, zidovudina 300 mg y abacavir 300 mg, administrados como comprimidos por separado, en lo que se refiere a los valores de AUCĄ y Cmáx. Los alimentos disminuyeron la velocidad de absorción de Tricivir® comprimidos (ligera disminución de la Cmáx (media 18-32%) y aumento en el valor de Tmáx (aproximadamente 1 hora), pero no el grado de absorción (AUCĄ). Estos cambios no se consideraron clínicamente significativos y no se recomiendan restricciones dietéticas para la administración de Tricivir®. Distribución: Los estudios intravenosos con lamivudina, abacavir y zidovudina mostraron que el volumen medio aparente de distribución es de 1.3; 0.8 y 1.6 l/kg, respectivamente. Lamivudina presenta una farmacocinética lineal a lo largo del intervalo de dosis terapéuticas y muestra una unión limitada a albúmina, la principal proteína del plasma (menos del 36% albúmina sérica in vitro). La unión de zidovudina con proteínas plasmáticas es de 34% a 38%. Los estudios in vitro de unión a proteínas plasmáticas indican que abacavir se une sólo en una proporción baja a moderada (aproximadamente 49%) a las proteínas del plasma humano a concentraciones terapéuticas. Esto indica una escasa probabilidad de interacciones con otros medicamentos por desplazamiento de la unión a proteínas plasmáticas. No se han previsto interacciones con medicamentos que impliquen el desplazamiento del sitio de unión con Tricivir® comprimidos. Los datos muestran que lamivudina, abacavir y zidovudina penetran en el sistema nervioso central (SNC) y llegan al líquido cefalorraquídeo (LCR). Las proporciones medias de concentración en LCR/concentración sérica de lamivudina y zidovudina después de 2-4 horas de la administración por vía oral fueron, aproximadamente, de 0.12 y 0.5 respectivamente. Se desconoce el grado real de la penetración de lamivudina y su relación con la eficacia clínica. Estudios realizados en pacientes infectados por VIH muestran una buena penetración de abacavir al líquido cefalorraquídeo (LCR) con una relación de AUC plasmática entre el 30 y el 44%. En la Fase I de un estudio farmacocinético, se investigó la penetración de abacavir al LCR después de la administración de 300 mg de abacavir 2 veces al día. La concentración media de abacavir alcanzada en el LCR después de 1.5 horas de la administración de la dosis fue de 0.14 µg/ml. En un estudio farmacocinético adicional en el que se administraron 600 mg 2 veces al día, la concentración de abacavir en el LCR incrementó con respecto al tiempo, de aproximadamente 0.13 µg/ml después de 0.5 a 1 hora de la administración de la dosis, a aproximadamente 0.74 µg/ml después de 3 a 4 horas. Mientras que los valores de las concentraciones máximas no se habían alcanzado después de 4 horas, los valores observados son 9 veces superiores a la CI50 de abacavir de 0.08 µg/ml ó 0.26 µM. Metabolismo: El metabolismo de lamivudina es una ruta menor de eliminación. El aclaramiento de lamivudina se realiza predominantemente mediante excreción renal del fármaco inalterado. La probabilidad de interacciones metabólicas con lamivudina es baja, debido al pequeńo grado de metabolismo hepático (5-10%) y a la escasa unión a proteínas plasmáticas. El principal metabolito de zidovudina en plasma y orina es el derivado 5'-glucurónido, originando que aproximadamente del 50 al 80% de la dosis administrada sea eliminada por excreción renal. Se ha identificado a la 3'-amino-3'-desoxitimidina (AMT) como un metabolito de zidovudina después de la dosificación por vía intravenosa. Abacavir se metaboliza principalmente en el hígado excretándose menos de un 2% de la dosis administrada por vía renal, como compuesto inalterado. Las principales vías metabólicas en el metabolismo del hombre son la de la alcohol deshidrogenasa y por glucuronidación o glucuronoconjugación para producir el ácido 5'-carboxílico y el 5'-glucurónido que representan alrededor del 66% de la dosis excretada en la orina. Eliminación: La vida media observada de la eliminación de lamivudina es de 5 a 7 horas. La depuración sistémica media de lamivudina es aproximadamente de 0.32 l/h/kg, con una depuración principalmente renal (mayor de 70%) a través del sistema de transporte catiónico del organismo. Estudios realizados en pacientes con alteración renal muestran que la eliminación de lamivudina se ve afectada por la disfunción renal. Se precisa de una reducción de dosis en pacientes con aclaramiento de creatinina menor o igual a 50 ml/min (véase Posología). En estudios realizados con zidovudina por vía intravenosa, el valor promedio de la vida media terminal en plasma fue de 1.1 horas y el aclaramiento sistémico medio fue de 1.6 l/h/kg. Se estima que el aclaramiento renal de zidovudina es 0.34 l/h/kg, indicando la existencia de filtración glomerular y secreción tubular activa en los rińones. Las concentraciones de zidovudina se incrementan en pacientes con deterioro renal avanzado. La vida media promedio del abacavir es de aproximadamente 1.5 horas. Tras la administración de múltiples dosis de 300 mg de abacavir 2 veces al día por vía oral, no se produce una acumulación significativa de abacavir. La eliminación de abacavir tiene lugar a través del metabolismo hepático con la posterior excreción de metabolitos principalmente en la orina. Los metabolitos y el abacavir inalterado representan un 83% de la dosis administrada de abacavir en la orina, siendo el resto eliminado en heces. Poblaciones especiales: Pacientes geriátricos: No se cuenta con datos farmacocinéticos para pacientes mayores de 65 ańos. Insuficiencia renal: Los estudios en pacientes con insuficiencia renal muestran que la eliminación de lamivudina se ve afectada por la disfunción renal, debido a la reducción en el clearance renal. Se requiere de reducción en la dosis en pacientes con clearance de creatinina menor de 50 ml/min. También se ha mostrado que las concentraciones de zidovudina aumentaron en pacientes con insuficiencia renal avanzada. Abacavir se metaboliza principalmente en el hígado excretándose aproximadamente un 2% de abacavir inalterado en orina. La farmacocinética de abacavir en pacientes con enfermedad renal en fase terminal es similar a la de pacientes con función renal normal. Dado que puede ser necesario ajustar la dosis de lamivudina y zidovudina, se recomienda la administración por separado de preparaciones de zidovudina, lamivudina y abacavir a pacientes con la función renal disminuida (clearance de creatinina menor o igual a 50 ml/min). Insuficiencia hepática: No se dispone de datos sobre el uso de Tricivir® comprimidos en pacientes con alteración hepática. El escaso número de datos de que se dispone en pacientes con cirrosis sugiere que la posible acumulación de zidovudina en pacientes con insuficiencia hepática puede ser debida a una disminución de la glucuronidación. Los datos obtenidos de pacientes con insuficiencia hepática moderada a severa, muestran que la farmacocinética de lamivudina no se ve afectada de forma significativa por la disfunción hepática. Abacavir se metaboliza principalmente en el hígado. Se realizaron estudios farmacocinéticos de abacavir en pacientes con insuficiencia hepática leve (Child-Pugh 5-6). Los resultados mostraron que hubo un incremento promedio de 1.89 veces en el área bajo la curva del abacavir y de 1.58 veces en la vida media de abacavir. Las áreas bajo la curva de los metabolitos no se vieron modificadas por la enfermedad hepática. Sin embargo, las tasas de formación y eliminación de estos, disminuyeron. Por lo tanto, se requiere de una reducción en la dosis de abacavir en pacientes con insuficiencia hepática leve. Se desconoce la farmacocinética de abacavir en pacientes con insuficiencia hepática de moderada a grave y, por lo tanto, está contraindicado el tratamiento con abacavir en ese grupo de pacientes (véase Contraindicaciones). Estudios clínicos: En pacientes no tratados previamente con fármacos antirretrovirales, la combinación triple de abacavir, lamivudina y zidovudina fue superior, en lo que se refiere a duración de la respuesta relativa a carga viral durante 48 semanas, a lamivudina y zidovudina. En una población de pacientes similar, se demostró la permanencia de la respuesta antiviral durante 120 semanas en, aproximadamente, el 70% de los pacientes. En pacientes no tratados previamente con fármacos antirretrovirales y que fueron tratados con una combinación de abacavir, lamivudina, zidovudina y efavirenz, la proporción de pacientes con carga viral indetectable (menos de 400 copias/ml) fue de aproximadamente el 90%, presentando un 80% menos de 50 copias/ml, al cabo de 24 semanas de tratamiento. Un estudio clínico aleatorio, doble ciego, con una duración de 48 semanas de tratamiento en pacientes no tratados previamente, la combinación de abacavir, lamivudina y zidovudina mostró un equivalente al efecto antiviral que se obtiene con la combinación de indinavir lamivudina y zidovudina. En un análisis secundario de pacientes con niveles de VIH-1 ARN en plasma al comienzo superiores a 100.000 copias por ml, los pacientes que recibieron la combinación que contenía indinavir mostraron una respuesta superior. Los pacientes con niveles de VIH-1 ARN en plasma al comienzo, inferiores a 100.000 copias por ml tuvieron una respuesta equivalente en ambos tratamientos. En pacientes con una carga viral baja al comienzo (menos de 5.000 copias/ml) y una exposición moderada al tratamiento antirretroviral, la incorporación del abacavir a un tratamiento previo incluyendo lamivudina y zidovudina produjo un impacto moderado sobre la carga viral a las 48 semanas. Actualmente, no se dispone de datos sobre el uso de Tricivir® en pacientes intensamente tratados previamente, en pacientes que han fracasado con otros tratamientos o en pacientes con enfermedad avanzada (células CD4 <50 células/mm3). El grado de beneficio de esta combinación de análogos de nucleósidos en pacientes sometidos previamente a un tratamiento intenso, dependerá de la naturaleza y duración del tratamiento anterior que podría haber dado lugar a variantes del VIH con resistencia cruzada a abacavir, lamivudina o zidovudina. Datos preclínicos de seguridad: No se dispone de datos preclínicos en animales sobre el tratamiento con la combinación de lamivudina, abacavir y zidovudina. Los efectos toxicológicos clínicos relevantes de los tres medicamentos son anemia, neutropenia y leucopenia. Mutagenicidad y carcinogenicidad: Ni lamivudina, abacavir ni zidovudina son mutagénicos en pruebas con bacterias pero, al igual que muchos análogos de nucleósidos, muestran actividad en pruebas in vitro sobre mamíferos tales como el ensayo de linfoma en ratón. Este hecho es concordante con la actividad conocida de otros análogos de nucleósidos. Lamivudina no ha mostrado ninguna actividad genotóxica en estudios in vivo. Sin embargo, zidovudina y abacavir mostraron efectos clastogénicos en las pruebas de micronúcleo en ratones. También se ha observado que los linfocitos sanguíneos periféricos provenientes de pacientes con SIDA que reciben tratamiento con zidovudina, que tienen un número más elevado de rupturas cromosómicas. Un estudio piloto demostró que la zidovudina se incorpora al AND nuclear de los linfocitos en adultos, incluyendo mujeres embarazadas, que están bajo tratamiento con zidovudina. No están claras las implicaciones clínicas de estos hallazgos. Lamivudina no mostró ningún potencial carcinogénico. En los estudios de carcinogenicidad oral con zidovudina en ratones y ratas, se observaron tumores en el epitelio vaginal, de aparición tardía. Un posterior estudio de carcinogenicidad intravaginal confirmó la hipótesis de que los tumores vaginales eran el resultado de la exposición local a largo plazo del epitelio vaginal del roedor a las altas concentraciones de zidovudina no metabolizado, en orina. No se observaron otros tumores relacionados con zidovudina en ninguno de los sexos de ninguna de las especies. Además, se han conducido dos estudios de carcinogenicidad transplacentaria en ratones. En un estudio del Instituto Nacional del Cáncer de los EE.UU., se administró zidovudina a ratonas preńadas, a las máximas dosis toleradas, durante los días 12 a 18 de gestación. Un ańo después del nacimiento, hubo un incremento en la incidencia de tumores en el pulmón, hígado y aparato reproductor femenino de la descendencia expuesta al nivel de dosis más elevado (420 mg/kg de peso corporal al final de la gestación). En el segundo estudio, se administró zidovudina a los ratones a dosis de hasta 40 mg/kg durante 24 meses, comenzando la exposición en el décimo día de gestación. Los hallazgos relacionados con el tratamiento se limitaron a tumores del epitelio vaginal de ocurrencia tardía, que se observó tienen una incidencia y tiempo de inicio similares al estudio de carcinogenicidad oral estándar. De este modo, el segundo estudio no proporcionó evidencia de que zidovudina actúe como un carcinógeno transplacentario. En los estudios de carcinogenicidad con abacavir administrado por vía oral en ratones y ratas se observó un incremento en la incidencia de tumores malignos y benignos. Los tumores malignos se presentaron en el prepucio en machos y en el clítoris en hembras de ambas especies y en el hígado, vejiga urinaria, nódulos linfáticos y en la subdermis de ratas hembras. La mayor parte de esos tumores se presentaron al nivel de dosis de abacavir más elevado de 330 mg/kg/día en ratones y 600 mg/kg/día en ratas. Los niveles de dosis fueron equivalentes a 24 a 32 veces la exposición sistémica esperada en humanos. La excepción fue el tumor del prepucio, el cual se presentó a una dosis de 110 mg/kg. Esto es equivalente a 6 veces la exposición sistémica en humanos. No existe una contraparte estructural de esta glándula en humanos. Mientras que se desconoce el potencial carcinogénico en humanos, estos datos sugieren que el riesgo carcinogénico en humanos se puede superarse por el beneficio clínico potencial. Toxicología en la reproducción: En los estudios de toxicología en la reproducción en animales, lamivudina, abacavir y zidovudina mostraron, todos, pasar a través de la placenta. Lamivudina no resultó ser teratogénico en los estudios con animales, pero existieron indicios de un incremento en las muertes embrionarias tempranas en el conejo con exposiciones sistémicas relativamente bajas, comparables a las alcanzadas en humanos. En las ratas no se observó un efecto similar, incluso con una exposición sistémica muy elevada. Zidovudina tuvo un efecto similar en ambas especies, aunque sólo a muy elevadas exposiciones sistémicas. A dosis tóxicas para la madre, zidovudina suministrado a ratas durante la organogénesis originó un aumento de la incidencia de malformaciones, aunque no se observó evidencia de anormalidades fetales a menores dosis. Se demostró la existencia de toxicidad debida a abacavir para el embrión en desarrollo y para el feto en ratas a dosis tóxicas maternas de 500 mg/kg/día y mayores. Esta dosis es equivalente a 32 a 35 veces la exposición terapéutica en humanos con base en el área bajo la curva. Los hallazgos incluyen el edema fetal, variaciones y malformaciones, reabsorciones, disminución en el peso corporal del feto y un incremento en el número de productos muertos al nacer. La dosis a la cual no se presentaron efectos sobre el desarrollo pre o post natal fue 160 mg/kg/día. Esta dosis es equivalente a una exposición de aproximadamente 10 veces a la del humano. No se observaron evidencias similares en conejos. Estudios de fertilidad realizados en la rata demostraron que abacavir, lamivudina y zidovudina carecen de efecto alguno sobre la fertilidad de machos o hembras. Toxicidad en dosis repetidas: Se observó una leve degeneración miocárdica en el corazón de ratones y ratas después de que se les administró abacavir por un período de 2 ańos. Las exposiciones sistémicas fueron equivalentes a 7 a 24 veces la exposición sistémica esperada en humanos. La relevancia clínica de este hallazgo no ha sido determinada.
|
|
Posología:
El tratamiento debe ser iniciado por un médico experimentado en el manejo de la infección por VIH. Cuando se indique la interrupción del tratamiento de uno de los principios activos del Tricivir® comprimidos, o cuando sea necesario reducir la dosis, se cuenta con preparados individuales de abacavir (Ziagen®0, lamivudina (Epivir) y zidovudina (Retrovir). Adultos y adolescentes: La dosis recomendada de Tricivir® comprimidos en adultos y adolescentes mayores de 12 ańos de edad es de 1 comprimido 2 veces al día. La formulación de Tricivir® puede tomarse con o sin alimentos. Tricivir® comprimidos no deberá administrarse a adolescentes o adultos que tengan un peso corporal menor a 40 kg debido a que el comprimido tiene una dosis fija y, por lo tanto, la dosis no puede ser reducida. Pacientes geriátricos: Actualmente, no se dispone de datos farmacocinéticos para pacientes mayores de 65 ańos. Se recomienda cuidado especial en este grupo de edad debido a los cambios asociados con la edad, tales como una disminución en la función renal y alteraciones en los parámetros hematológicos. Insuficiencia renal: Una reducción en la dosis de lamivudina o zidovudina puede ser necesaria en pacientes con insuficiencia renal. Por lo tanto, se recomienda administrar preparados individuales de abacavir, lamivudina y zidovudina a pacientes con función renal disminuida (clearence de creatinina inferior a 50 ml/min) (véase Propiedades farmacocinéticas). Insuficiencia hepática: Tricivir® comprimidos está contraindicado para administrarse en pacientes con insuficiencia hepática (véase Contraindicaciones y Propiedades farmacocinéticas). Ajustes a la dosificación en pacientes con reacciones hematológicas adversas: Puede que sea necesario ajustar la dosificación de zidovudina si el nivel de hemoglobina baja a menos de 9 g/dl ó 5.59 mmol/l o si el recuento de neutrófilos baja a menos de 1.0 x 10 ł 9/l (véase Contraindicaciones y Advertencias y precauciones especiales de empleo). Por lo tanto, deberán administrarse preparados de abacavir, zidovudina y lamivudina por separado.
|
|
Modo de Empleo:
No hay requisitos especiales.
|
|
Efectos Colaterales:
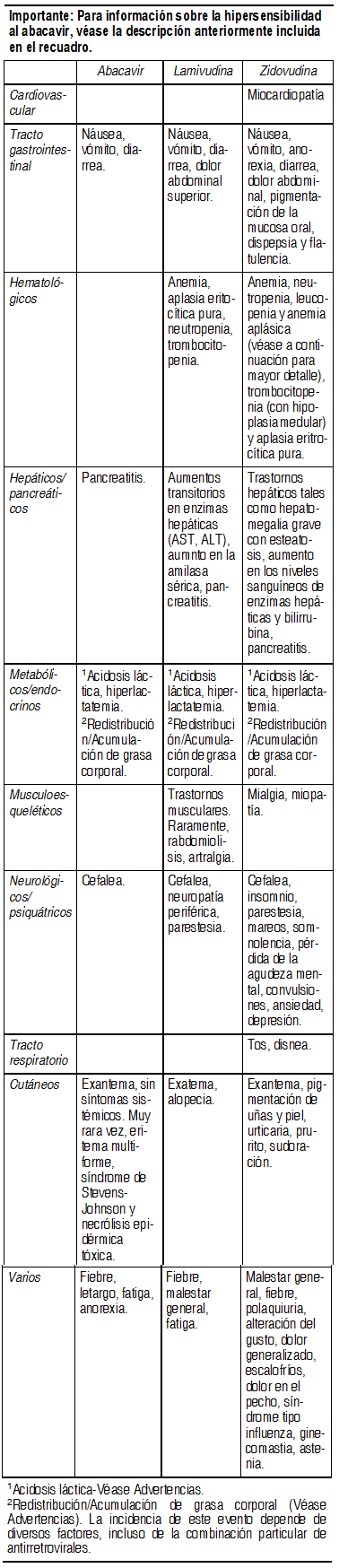
Tricivir® comprimidos contiene abacavir, lamivudina y zidovudina. Por lo tanto, puede esperarse la aparición de las reacciones adversas asociadas con estos compuestos, que se presentan en la Tabla 1 a continuación, después de iniciar un tratamiento con Tricivir® comprimidos. En muchos casos no queda claro si estos eventos están relacionados con el principio activo, con la amplia gama de medicamentos utilizados en el control de la enfermedad causada por el VIH o si son consecuencia del proceso de la enfermedad subyacente. La estimación del perfil de seguridad del Tricivir® comprimidos en estudios clínicos no se encuentra todavía disponible. Hipersensibilidad al abacavir: (también véase Advertencias). En general, en estudios clínicos realizados antes de la introducción del procedimiento de detección del alelo HLA-B*5701, aproximadamente el 5% de los individuos que recibieron abacavir desarrollaron una reacción de hipersensibilidad, la cual en raros casos produjo la muerte. Estas reacciones se caracterizan por la aparición de síntomas compromiso multisistémico o multiorgánico. Casi todos los pacientes que desarrollen reacciones de hipersensibilidad presentarán fiebre y/o exantema (normalmente maculopapular o urticariforme) como parte del síndrome, no obstante han tenido lugar reacciones de hipersensibilidad sin exantema o fiebre. Pueden aparecer síntomas en cualquier momento del tratamiento con abacavir, pero normalmente aparecen dentro de las 6 primeras semanas de inicio del tratamiento (mediana del tiempo de inicio de 11 días). Los signos y los síntomas asociados con la reacción de hipersensibilidad se encuentran relacionados a continuación. Los signos y los síntomas reportados al menos por el 10% de los pacientes que presentaron una reacción de hipersensibilidad se encuentran en un texto marcado en negrita. Cutáneos: Exantema (generalmente, maculo-papuloso o urticaria). Tracto gastrointestinal: Náusea, vómito, diarrea, dolor abdominal, úlceras bucales. Tracto respiratorio: Disnea, tos, odinofagia, síndrome de distrés respiratorio del adulto, insuficiencia respiratoria. Varios: Fiebre, fatiga, malestar general, edema, linfoadenopatia, hipotensión, conjuntivitis, anafilaxia. Neurológicos/psiquiátricos: Cefalea, parestesia. Hematológicos: Linfopenia. Hepáticos/pancreáticos: Pruebas de la función hepática elevada, insuficiencia hepática. Musculoesqueléticos: Mialgia, miolisis poco frecuente, artralgia, creatinin fosfoquinasa elevada. Urológicos: Creatinina elevada, insuficiencia renal. Inicialmente, en el caso de algunos pacientes que presentaron reacción de hipersensibilidad, se pensó que tenían una enfermedad respiratoria (neumonía, bronquitis, faringitis), una afección de tipo gripal, gastroenteritis o reacciones a otros medicamentos. El retraso en el diagnóstico de una reacción de hipersensibilidad ha ocasionado que se continúe la administración de abacavir o bien que se vuelva a administrar, induciendo al paciente a una reacción de hipersensibilidad más severa o a la muerte. Por lo tanto, en aquellos pacientes que presenten síntomas de estas enfermedades se debe de considerar cuidadosamente un diagnóstico de reacción de hipersensibilidad. En el caso de que no sea posible descartar una reacción de hipersensibilidad, no se deberá reiniciar el tratamiento con Tricivir® comprimidos o con ningún medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®). Los síntomas que se encuentran relacionados con esta reacción de hipersensibilidad empeoran conforme se continúa con el tratamiento y generalmente se solucionan al interrumpir la administración de abacavir. El reinicio del tratamiento con abacavir después de una reacción de hipersensibilidad conduce a que rápidamente, en cuestión de horas, vuelvan a aparecer los síntomas. Esta recurrencia de la reacción de hipersensibilidad puede ser más grave que la que se experimentó en la presentación inicial y puede incluir hipotensión con riesgo para la vida e incluso la muerte. Independientemente de su estatus de HLA-B*5701, los pacientes que desarrollen esta reacción de hipersensibilidad deberán interrumpir la administración de Tricivir®® comprimidos y nunca deberán reiniciar el tratamiento con Tricivir® comprimidos o con ningún medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®). En muy raras ocasiones, se han notificado reacciones de hipersensibilidad después de la reintroducción del tratamiento con abacavir, en pacientes cuya interrupción fue precedida por un solo síntoma clave de hipersensibilidad (exantema fiebre, malestar general/fatiga, síntomas gastrointestinales o un síntoma respiratorio). En muy raras ocasiones, se han reportado reacciones de hipersensibilidad en pacientes en quienes se ha reiniciado la terapia y no tienen antecedentes de síntomas de una reacción de hipersensibilidad.
Ver Tabla
Efectos adversos con abacavir: Muchos de los efectos adversos listados anteriormente para el abacavir (náuseas, vómitos, diarrea, fiebre, fatiga, exantema) comúnmente ocurren como parte de la reacción de hipersensibilidad al abacavir. Por tanto, se debe evaluar cuidadosamente a los pacientes que presenten cualquiera de estos síntomas para descartar la presencia de esta reacción de hipersensibilidad. Si se ha suspendido la terapia con Tricivir® comprimidos en alguno paciente debido a que experimentaron algunos de estos síntomas y se toma la decisión de reiniciar la terapia con este fármaco, esto sólo debe hacerse bajo supervisión médica directa (véase Consideraciones especiales a tomar después de suspender una terapia con Tricivir®). Reacciones adversas de tipo hematológico relacionadas con zidovudina: Anemia (la cual puede requerir transfusiones), neutropenia, leucopenia y anemia aplásica, se produjeron con mayor frecuencia a dosis más elevadas (1.200-1.500 mg/día) y en pacientes en etapa avanzada de la enfermedad por VIH (especialmente cuando existe una deficiente reserva de médula ósea antes del tratamiento) y, particularmente, en pacientes con recuentos de células CD4 inferiores a 100/mm3. Puede llegar a requerirse una reducción en la dosis o la interrupción de la terapia (véase Advertencias y Precauciones). También aumentó la incidencia de neutropenia en pacientes con recuentos bajos de neutrófilos y con bajos niveles de hemoglobina y de vitamina B12 sérica al inicio de la terapia con zidovudina.
|
|
Contraindicaciones:
El uso de Tricivir® comprimidos está contraindicado en pacientes con hipersensibilidad conocida al Tricivir® o a cualquiera de sus componentes (abacavir, lamivudina o zidovudina), o a cualquier excipiente de los comprimidos de Tricivir®. Tricivir® comprimidos está contraindicado en pacientes con insuficiencia hepática. Debido al principio activo de la zidovudina, el uso de Tricivir® comprimidos está contraindicado en pacientes con recuento de neutrófilos anormalmente bajos (menos de 0.75 x 109/l) o niveles de hemoglobina anormalmente bajos (<7.5 g/dl ó 4.65 mmol/l) (véase Advertencias).
|
|
Advertencias:
Hipersensibilidad al abacavir: (ver también Efectos colaterales). En general, en estudios clínicos realizados antes de la introducción del procedimiento de detección del alelo HLA-B*5701, aproximadamente el 5% de los individuos que recibieron abacavir desarrollaron una reacción de hipersensibilidad, la cual en raros casos produjo la muerte. Factores de riesgo: Los estudios han demostrado que la presencia del alelo HLA B*5701 está asociado con un aumento significativo en el riesgo de desarrollar una reacción de hipersensibilidad al abacavir. En el estudio prospectivo CNA106030 (PREDICT-1), el uso de un método de detección del alelo HLA B*5701 previo a la terapia , y la prevención subsiguiente de la administración de abacavir a pacientes con este alelo, redujo la tasa de incidencia de reacciones de hipersensibilidad clínicamente sospechosas al abacavir, de 7.8% (66 de 847) a 3.4% (27 de 803) (p<0.0001), y la tasa de incidencia de reacciones de hipersensibilidad confirmadas mediante la prueba del parche cutáneo, de 2.7% (23 de 842) a 0.0% (0 de 802) (p<0.0001). Con base en este estudio, se estima que de 48% a 61% de los pacientes que presentan el alelo HLA B*5701 desarrollará alguna reacción de hipersensibilidad durante el ciclo terapéutico con abacavir, en comparación con 0% a 4% de los pacientes que no presentan el alelo HLA B*5701. Los médicos deberían contemplar la utilización de métodos de detección del alelo HLA B*5701 en cualquier paciente infectado por VIH que no haya sido expuesto previamente al abacavir. Se recomienda realizar el screening para el alelo HLA B*5701antes de reiniciar el tratamiento con abacavir en pacientes con estatus de HLA-B*5701 desconocido, aunque hayan tolerado previamente el abacavir (véase "Consideraciones especiales después de la interrupción de una terapia con abacavir"). No se recomienda el uso de abacavir en pacientes que sean conocidos portadores del alelo HLA B*5701, por lo cual deberá contemplarse su uso sólo bajo circunstancias excepcionales donde el beneficio potencial exceda al riesgo, y bajo una estrecha supervisión médica. En cualquier paciente tratado con abacavir, el diagnóstico clínico de una reacción de hipersensibilidad sospechosa deberá seguir siendo la base en la toma de decisiones clínicas. Aún en ausencia del alelo HLA B*5701, es importante suspender la administración del abacavir, y no volver a exponer al paciente a este fármaco, si no es posible descartar una reacción de hipersensibilidad por motivos clínicos, debido al riesgo de que se presente una reacción severa, incluso mortal. Descripción clínica: Las reacciones de hipersensibilidad se caracterizan por la aparición de síntomas que indican que varios órganos se encuentran afectados. En la mayoría de los pacientes se presenta fiebre y/o exantema como parte del síndrome. Otros de los síntomas de la reacción de hipersensibilidad pueden incluir fatiga, malestar general, síntomas gastrointestinales como náusea, vómito, diarrea o dolor abdominal y signos y síntomas respiratorios como disnea, odinofagia, tos y hallazgos anormales en la radiografía de tórax (predominantemente infiltrados, los cuales pueden ser localizados). Los síntomas de esta reacción de hipersensibilidad pueden presentarse en cualquier momento durante el tratamiento con abacavir, pero por lo general se presentan dentro de las primeras 6 semanas del tratamiento. Los síntomas empeoran al continuar con la administración y se puede poner en peligro la vida del paciente. Generalmente, estos síntomas se resuelven al suspender la administración de abacavir. Manejo clínico: Independientemente de su estatus de HLA-B*5701, cualquier paciente que desarrolla signos o síntomas de hipersensibilidad debe ponerse en contacto con su médico inmediatamente para ser evaluados. Si se diagnostica alguna reacción de hipersensibilidad deben interrumpir la administración de Tricivir® comprimidos inmediatamente. Nunca se debe reiniciar el tratamiento con Tricivir® comprimidos o con cualquier otro medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®), en pacientes que han presentado una reacción de hipersensibilidad, debido a que, en cuestión de horas, pueden volver a aparecer los síntomas con una recurrencia más grave que la inicial, incluyendo hipotensión severa e incluso la muerte. Para evitar un retraso en el diagnóstico y reducir al mínimo el riesgo de una reacción de hipersensibilidad que ponga en peligro la vida del paciente, debe interrumpirse permanentemente el tratamiento con Tricivir® comprimidos en caso de que no pueda descartarse la hipersensibilidad, incluso cuando otros diagnósticos sean posibles (enfermedades respiratorias, cuadro pseudogripal, gastroenteritis o reacciones a otros medicamentos). No deberá reiniciarse el tratamiento con Tricivir® comprimidos o con cualquier otro medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®), aún en el caso de que existan síntomas de reincidencia posteriores a una terapia distinta con medicamentos alternativos. En el envase de Tricivir® comprimidos se incluye una tarjeta de alerta para el paciente con indicaciones acerca de la reacción de hipersensibilidad. Consideraciones especiales después de la interrupción del tratamiento con Tricivir® comprimidos: Independiente del estatus del HLA-B*5701 de un paciente, si por cualquier razón se ha interrumpido el tratamiento con Tricivir® comprimidos y se considera la posibilidad de reiniciar el tratamiento, se debe determinar la causa de la interrupción para asegurarse que el paciente no tuvo algún síntoma de una reacción de hipersensibilidad antes de interrumpirlo. Si no puede descartarse la posibilidad de una reacción de hipersensibilidad, no debe reiniciarse el tratamiento con Tricivir® comprimidos ni ningún otro medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®). Con poca frecuencia se han notificado reacciones de hipersensibilidad después de la introducción del tratamiento con abacavir, en pacientes cuya interrupción fue precedida por un solo síntoma clave de hipersensibilidad (erupción cutánea, fiebre, malestar general/fatiga, síntomas gastrointestinales o un síntoma respiratorio). Si se decide reiniciar el tratamiento con Tricivir® comprimidos en esa clase de pacientes, este debe realizarse solamente bajo una supervisión médica directa. En muy raras ocasiones se han reportado reacciones de hipersensibilidad en pacientes en quienes se ha reiniciado la terapia y no tienen antecedentes de síntomas de una reacción de hipersensibilidad. Si se decide reiniciar el tratamiento con Tricivir® comprimidos en esa clase de pacientes, este debe realizarse solamente si el paciente o sus familiares tendrán un rápido acceso a la asistencia médica. Se recomienda realizar la detección del alelo HLA-B*5701 antes de reiniciar el tratamiento con abacavir en pacientes con estatus de HLA-B*5701 desconocido, y que hayan tolerado previamente el abacavir. No se recomienda reiniciar el tratamiento con abacavir en aquellos pacientes con resultados positivos para la prueba de detección del alelo HLA-B*5701, y sólo se debe contemplar en circunstancias excepcionales donde el beneficio potencial exceda el riesgo, y con una estrecha supervisión médica. Información esencial para el paciente: Quien prescribe este medicamento debe asegurarse de que los pacientes estén completamente informados con respecto a la siguiente información sobre la reacción de hipersensibilidad: Los pacientes deben estar conscientes de la posibilidad de presentar alguna reacción de hipersensibilidad al abacavir, la cual puede dar lugar a una reacción potencialmente mortal e incluso a la muerte y que existe un mayor riesgo de que desarrollen alguna reacción de hipersensibilidad si son HLA-B*5701-positivos. Además, se debe informar a los pacientes que las personas HLA-B*5701-negativas también pueden experimentar reacciones de hipersensibilidad al abacavir. Por lo tanto, cualquier paciente que desarrolle signos o síntomas consistentes con una posible reacción de hipersensibilidad al abacavir, debe contactar inmediatamente a su médico. Se deberá recordar a los pacientes hipersensibles al abacavir que nunca más deberán tomar Tricivir® comprimidos, ni cualquier otro medicamento que contenga abacavir (por ejemplo: Ziagen®, Kivexa®), independiente de su estatus de HLA-B*5701. Con el fin de evitar que reinicien el tratamiento con Tricivir® comprimidos, deberá pedirse a los pacientes que hayan experimentado una reacción de hipersensibilidad que devuelvan los comprimidos de Tricivir® comprimidos restantes a la farmacia. Se debe aconsejar a aquellos pacientes que por cualquier motivo hayan interrumpido el tratamiento con Tricivir® comprimidos y, especialmente cuando esto se haya debido a posibles reacciones adversas o enfermedad, que se pongan en contacto con su médico antes de reiniciarlo. Se debe recordar a cada paciente que lea la hoja impresa incluida en el envase de Tricivir® comprimidos. Se les debe recordar también la importancia de sacar la Tarjeta de Alerta incluida en el envase y llevarla consigo siempre. Acidosis láctica/hepatomegalia grave con esteatosis: Se ha reportado acidosis láctica y hepatomegalia grave con esteatosis, incluso casos fatales, con el uso de antirretrovirales análogos de nucleósidos, ya sea solos o en combinaciones, entre los que se incluye a abacavir, lamivudina y zidovudina. La mayoría de estos casos se ha presentado en mujeres. Entre las características clínicas que pueden indicar el desarrollo de acidosis láctica se incluyen: debilidad generalizada, anorexia y pérdida de peso corporal repentina e inexplicable, síntomas gastrointestinales y síntomas respiratorios (disnea y taquipnea). Debe tenerse precaución cuando se administra Tricivir® comprimidos a cualquier paciente y, en particular, a los que se sabe tienen factores de riesgo de enfermedad hepática. Debe suspenderse el tratamiento con Tricivir® comprimidos en cualquier paciente que desarrolle evidencia clínica o de laboratorio que sugiera acidosis láctica o hepatotoxicidad (que pueden incluir hepatomegalia y esteatosis, aún en ausencia de elevaciones notables en los niveles de aminotransferasas). Redistribución de lípidos: Se ha observado una redistribución/acumulación de grasa corporal, incluyendo obesidad central, aumento de la grasa dorsocervical (cuello de búfalo), adelgazamiento periférico, adelgazamiento facial, crecimiento mamario, aumento en los niveles de lípidos séricos y de glucosa sanguínea, ya sea como eventos aislados o en conjunto, en algunos de los pacientes que reciben terapia antirretroviral de combinación (véanse Efectos colaterales). Aunque todos los medicamentos de la clases de los inhibidores de la proteasa y de los inhibidores de la transcriptasa inversa de tipo nucleosídico han sido asociados con al menos alguno de estos eventos adversos específicos, relacionados con un síndrome general conocido comúnmente como lipodistrofia, los datos indican que existen diferencias en los riesgos que presentan cada uno de los miembros de las respectivas clases terapéuticas. Además, el síndrome de lipodistrofia tiene una etiología multifactorial; por ejemplo, en el estado de la enfermedad ocasionada por el VIH, tanto la edad del paciente como la duración del tratamiento antirretrovírico juegan un papel importante y posiblemente sinérgico. Actualmente se desconocen las consecuencias a largo plazo de estos eventos. El examen clínico debe incluir una evaluación de los signos físicos de redistribución de lípidos. Se debe considerar la medición de los niveles de lípidos séricos y glucosa en la sangre. Los trastornos en los niveles de lípidos deben controlarse según sea adecuado desde el punto de vista clínico. Reacciones hematológicas adversas: Puede esperarse que se presente anemia, neutropenia y leucopenia (normalmente en forma secundaria a la neutropenia) en los pacientes que están recibiendo zidovudina. Estas se presentaron con más frecuencia a altas dosis de zidovudina (1.200-1.500 mg/día) y en pacientes con deficiente reserva de médula ósea antes del tratamiento, particularmente con avanzada enfermedad por VIH. Por lo tanto, los parámetros hematológicos de los pacientes que reciben Tricivir® comprimidos deben ser cuidadosamente supervisados (véase Contraindicaciones). Estos efectos hematológicos no se observan normalmente antes de 4 a 6 semanas de tratamiento. Para pacientes con un avanzado estado sintomático de la enfermedad por VIH, se recomienda en general que las pruebas sanguíneas se realicen al menos cada 2 semanas durante los primeros 3 meses de tratamiento y, posteriormente, al menos 1 vez al mes. En pacientes que presentan un estado temprano de la enfermedad por VIH, son poco frecuentes las reacciones hematológicas adversas. Dependiendo del estado general del paciente, los análisis de sangre pueden efectuarse con menor frecuencia, por ejemplo, cada 1 a 3 meses. Puede requerirse de un ajuste adicional en la dosis de zidovudina en caso de que se presente anemia grave o mielosupresión durante el tratamiento con Tricivir® comprimidos, o en pacientes con compromiso previo de la médula ósea, por ejemplo con niveles de hemoglobina <9 g/dl (5.59 mmol/l) o recuento de neutrófilos <1.0 x 109/l (véase Posología). Dado que no es posible ajustar la dosis de Tricivir® comprimidos, deben utilizarse preparaciones separadas de zidovudina, abacavir y lamivudina. Pancreatitis: Muy rara vez se han presentado casos de pancreatitis en pacientes tratados con abacavir, lamivudina y zidovudina. Sin embargo, no está claro si estos casos se debieron al tratamiento con los medicamentos o a la enfermedad por VIH ya existente. El tratamiento con Tricivir® comprimidos deberá interrumpirse inmediatamente si aparecen signos clínicos, síntomas o anormalidades en los resultados de laboratorio indicativos de pancreatitis. Pacientes coinfectados con el virus de hepatitis B: Las pruebas clínicas y el empleo comercial de lamivudina han mostrado que algunos pacientes con enfermedad crónica por el virus de hepatitis B (VHB) pueden experimentar evidencia clínica o de laboratorio de hepatitis recurrente al discontinuar el empleo de la lamivudina, lo que tiene consecuencias más graves en pacientes con hepatopatía descompensada. En caso de que se interrumpa la administración de Tricivir® comprimidos en pacientes coinfectados con el virus de hepatitis B, debe considerarse la supervisión periódica con pruebas del funcionamiento hepático y con marcadores de replicación del VHB. Pacientes coinfectados por el virus de hepatitis C: Al administrar zidovudina como parte del régimen utilizado en el tratamiento del VIH, se han notificado casos de exacerbación de anemia ocasionada por la administración de ribavirina. Sin embargo, aún no se ha dilucidado el mecanismo exacto. Por tanto, no se recomienda administrar ribavirina y zidovudina de manera concomitante y, si esto ya se encuentra establecido, se debe contemplar un reemplazo de la zidovudina en una terapia antirretroviral (TARV) de combinación ya establecido. Esto es particularmente importante en pacientes con antecedentes conocidos de anemia inducida por la administración de zidovudina. Síndrome de reconstitución inmunológica: En aquellos pacientes infectados por el VIH, que presentan una deficiencia inmunitaria de grado severo al momento de iniciar la terapia antirretroviral (TARV), puede ocurrir una reacción inflamatoria, a infecciones oportunistas asintomáticas o residuales, que ocasionen serios trastornos clínicos o un agravamiento de los síntomas. Normalmente estas reacciones se observan dentro de las primeras semanas o meses posteriores al inicio de la TARV. Ejemplos importantes son la retinitis citomegalovírica, infecciones micobacterianas generalizadas o focales, o ambas, así como neumonía ocasionada por cepas de Pneumocystis jiroveci (P. carinii). Se debe evaluar, sin demora alguna, cualquier síntoma inflamatorio que se presente y, cuando sea necesario, iniciar un tratamiento. Se ha reportado la aparición de enfermedades autoinmunes (tales como enfermedad de Graves, polimiositis y síndrome de Guillain-Barre) en el contexto de la reconstitución inmunitaria; sin embargo, el tiempo de aparición es mas variable, y puede ocurrir muchos meses después del inicio del tratamiento y algunas veces pueden ser de presentación atípica. Infecciones oportunistas: Los pacientes tratados con Tricivir® comprimidos o con cualquier otra terapia antirretroviral pudieran seguir desarrollando infecciones oportunistas u otras complicaciones de la infección por VIH. Por lo tanto, los pacientes deben permanecer bajo estrecha observación clínica de médicos experimentados en el tratamiento de estas enfermedades asociadas con la infección por VIH. Transmisión de la infección: Deberá advertirse a los pacientes que no se ha demostrado que la actual terapia antirretroviral, incluyendo el tratamiento con Tricivir® comprimidos, evite el riesgo de transmitir el VIH a otras personas por medio del contacto sexual o la contaminación sanguínea. Por lo tanto, deben seguirse tomando las precauciones apropiadas. Coadministración de otros medicamentos: Deberá advertirse a los pacientes sobre el uso concomitante de medicamentos autoadministrados (véase Interacciones). Ajustes en la dosis: En los casos donde sea necesario ajustar la dosis, se deberán administrar preparaciones separadas de abacavir, lamivudina y zidovudina. En estos casos, el médico deberá referirse a la información individual para prescribir de cada uno de estos medicamentos. Infarto de miocardio: En un estudio epidemiológico de carácter prospectivo y observacional, diseńado para investigar el índice de casos de infartos de miocardio en pacientes bajo terapia antirretroviral de combinación de fármacos, el uso de abacavir dentro de los seis meses previos estuvo correlacionado con un incremento en el riesgo de desarrollar infarto de miocardio. En un análisis global de estudios clínicos patrocinados por GSK, no se observó riesgo excedente alguno de desarrollar infarto de miocardio con el uso de abacavir. No existe un mecanismo biológico conocido que explique un incremento potencial. Todos los datos disponibles a partir de cohortes observacionales y estudios clínicos controlados no son concluyentes en lo que respecta a la relación entre el tratamiento con abacavir y el riesgo de desarrollar infarto de miocardio. Como medida precautoria, se debería considerar el riesgo subyacente de cardiopatía coronaria al prescribir terapias antirretrovirales, incluyendo abacavir, y tomar las medidas necesarias para minimizar todos los factores de riesgo modificables (por ej., hipertensión, hiperlipidemia, diabetes mellitus y tabaquismo).
|
|
Precauciones:
Embarazo y lactancia: Fertilidad: No hay datos del efecto de abacavir, lamivudina o zidovudina sobre la fertilidad de la mujer. En los hombres, se ha mostrado que zidovudina no tiene efecto sobre la cuenta espermática, la morfología o la motilidad. Embarazo: No se ha establecido la seguridad en el empleo de Tricivir® comprimidos durante el embarazo humano. Los estudios de reproducción realizados en animales con lamivudina, abacavir y zidovudina han sido asociados con los hallazgos obtenidos (véase Datos preclínicos de seguridad). Por lo tanto, la administración de Tricivir® comprimidos durante el embarazo solamente deberá ser considerada en el caso de que el beneficio a la madre sobrepase el posible riesgo al feto. Ha habido comunicaciones de aumentos leves y transitorios en los niveles lácticos en el suero, los cuales pueden deberse a una disfunción mitocondrial, en neonatos y lactantes expuestos en el útero o periparto a los inhibidores nucleosídicos de la transcriptasa inversa (INTIs). Se desconoce la relevancia clínica de los aumentos transitorios en los niveles de lactato en el suero. También ha habido comunicaciones muy raras de retrasos en el desarrollo, convulsiones y otros padecimientos neurológicos. Sin embargo, no se ha establecido una relación causal entre estos eventos y la exposición a INTIs en el útero o periparto. Estos hallazgos no influyen en las recomendaciones actuales sobre el uso de terapia antirretroviral en mujeres embarazadas para prevenir la transmisión vertical del VIH. Lactancia: Los expertos en salud recomiendan que, cuando sea posible, las mujeres infectadas con VIH no amamanten a sus bebés bajo ninguna circunstancia, con el fin de evitar la transmisión del VIH. Tanto lamivudina como zidovudina se excretan en la leche humana a concentraciones similares a las que se encuentran en el suero. Es de esperar que el abacavir se excrete también en la leche humana, aunque este hecho no se ha confirmado. Se recomienda que las madres que tomen Tricivir® comprimidos no amamanten a sus hijos. Efectos sobre la capacidad de conducir y operar maquinaria: No se han realizado estudios para investigar el efecto de Tricivir® comprimidos o de sus componentes activos (abacavir, lamivudina y zidovudina) sobre la capacidad para conducir vehículos o para operar maquinaria. Además, el efecto perjudicial sobre estas actividades no puede pronosticarse a partir de la farmacología de las sustancias activas. Debe tenerse en cuenta el estado clínico del paciente y el perfil del suceso adverso del Tricivir® comprimidos cuando se considere la capacidad del paciente para conducir vehículos u operar maquinaria.
|
|
Interacciones Medicamentosas:
Estudios clínicos han demostrado que no existen interacciones clínicamente significativas entre abacavir, zidovudina y lamivudina. Dado que Tricivir® comprimidos contiene abacavir, lamivudina y zidovudina, cualquier interacción que haya sido identificada individualmente con estos agentes puede presentarse con Tricivir® comprimidos. La enumeración de interacciones que se da a continuación no debe considerarse como exhaustiva, sino que es sólo representativa de las clases de medicamentos con los que debe tenerse precaución. Interacciones relativas al abacavir: En función de los resultados de las pruebas in vitro y de las principales rutas metabólicas conocidas de abacavir, es baja la posibilidad de que tengan lugar interacciones con otros medicamentos mediadas por el citocromo P450, en las que esté implicado abacavir. El abacavir no ha mostrado la posibilidad de inhibir el metabolismo mediado por la enzima citocromo P450 3A4. También se ha demostrado in vitro que el abacavir no inhibe las enzimas CYP 3A4, CYP 2C9 o CYP 2D6. No se ha observado la inducción del metabolismo hepático en estudios clínicos. Por lo tanto, la posibilidad de que existan interacciones con inhibidores antirretrovirales de la proteasa y otros medicamentos metabolizados por las principales enzimas P450, es mínima. Etanol: El metabolismo de abacavir se ve alterado por la administración concomitante de etanol originándose un incremento de aproximadamente un 41% en el área bajo la curva de abacavir. Estos hallazgos no se consideran clínicamente significativos. El abacavir carece de efecto sobre el metabolismo de etanol. Metadona: En un estudio farmacocinético, la coadministración de 600 mg de abacavir 2 veces al día con metadona dio lugar a una reducción del 35% en la Cmáx del abacavir y a un retraso de 1 hora en el valor de tmáx, permaneciendo inalterada el área bajo la curva. Los cambios en la farmacocinética de abacavir no se consideran relevantes desde el punto de vista clínico. En este estudio el abacavir incrementó el promedio de depuración sistémico de metadona en un 22%. Este cambio no se considera importante desde el punto de vista clínico para la mayoría de los pacientes aunque, en ocasiones, se requiere volver a determinar la dosis de metadona. Retinoides: Los compuestos retinoides como la isotretinoina, se eliminan mediante el alcohol deshidrogenasa. Es posible la interacción con abacavir pero no ha sido estudiada. Interacciones relativas a lamivudina: Es baja la probabilidad de interacciones metabólicas con lamivudina, debido al limitado metabolismo y unión de la proteína plasmática y a la casi completa eliminación renal de la lamivudina intacto. Deberá considerarse la posibilidad de interacciones con otros medicamentos administrados al mismo tiempo que Tricivir® comprimidos, particularmente cuando la principal vía de eliminación sea la secreción renal activa. Trimetoprim: La administración de trimetoprim/sulfametoxazol 160 mg/800 mg (cotrimoxazol) provoca un aumento del 40% en la exposición a la lamivudina, debido al componente trimetoprim. Sin embargo, a menos que el paciente tenga alguna insuficiencia renal, no es necesario ajustar la dosis de lamivudina (véase Posología). Lamivudina no tiene efecto sobre la farmacocinética del trimetoprim o del sulfametoxazol. No se ha estudiado el efecto de la coadministración de lamivudina con las altas dosis de cotrimoxazol utilizadas para el tratamiento de neumonía por Pneumocystis carinii y toxoplasmosis. Zalcitabina: Lamivudina puede inhibir la fosforilación intracelular de zalcitabina cuando los dos medicamentos se emplean en forma concurrente. Por lo tanto, no se recomienda el empleo de Tricivir® en combinación con zalcitabina. Interacciones relativas a zidovudina: La zidovudina se elimina principalmente por conjugación hepática como un metabolito glucuronizado inactivo. Los medicamentos que se eliminan principalmente mediante el metabolismo hepático, especialmente a través de la glucuronización, pueden tener el potencial de inhibir el metabolismo de zidovudina. Atovacuona: La zidovudina no parece afectar el perfil farmacocinético de la atovacuona. Sin embargo, la información farmacocinética ha demostrado que la atovacuona aparentemente disminuye la velocidad metabólica de la zidovudina a su metabolito glucurónido (el ABC en estado estacionario de la zodovudina experimentó un aumento de 33%, mientras las concentraciones plasmáticas máximas del glucurónido disminuyeron 19%). Al administrar dosis de zidovudina de 500 ó 600 mg/día, parecería improbable que un ciclo terapéutico concomitante de 3 semanas de duración, con atovacuona administrada en el tratamiento de la NPC (neumonía por P. carinii) aguda, sea capaz de producir un aumento en la tasa de incidencia de efectos adversos atribuibles a las elevadas concentraciones plasmáticas de zidovudina. Se deberá tener más cuidado al vigilar a pacientes que reciban tratamiento con atovacuona durante un periodo prolongado. Claritromicina: Los comprimidos de claritromicina reducen el grado de absorción de la zidovudina. Esto puede evitarse al separar la administración de zidovudina y claritromicina por un intervalo de cuando menos 2 horas. Lamivudina: La coadministración de zidovudina con lamivudina resulta en un aumento del 13% en la exposición a zidovudina y un aumento de 28% en los niveles plasmáticos máximos. Sin embargo, la exposición general (área bajo la curva) no se altera en forma significativa. Este incremento no se considera de importancia significativa para la seguridad del paciente y por lo tanto, no se requiere de un ajuste en la dosis. Zidovudina no tiene efecto sobre la farmacocinética de lamivudina. Fenitoína: Se ha reportado que los niveles de fenitoína en sangre son bajos en algunos pacientes que reciben zidovudina, mientras que en un paciente se observó un nivel elevado. Estas observaciones indican que las concentraciones de fenitoína deben ser cuidadosamente supervisadas en los pacientes que reciben Tricivir® comprimidos y fenitoína. Probenecid: Algunos datos limitados sugieren que probenecid aumenta la vida media y el área bajo la curva de concentración plasmática de zidovudina, al disminuir la glucuronoconjugación. La excreción renal del glucurónido (y posiblemente del propio zidovudina) se reduce en presencia de probenecid. Rifampicina: Ciertos datos limitados sugieren que la coadministración de zidovudina y rifampicina disminuye el área bajo la curva de zidovudina en 48% ± 34%. Sin embargo, se desconoce su importancia clínica. Estavudina: La zidovudina puede inhibir la fosforilación intracelular de estavudina cuando los dos medicamentos se emplean en forma concurrente. Por lo tanto, no se recomienda la administración de estavudina en combinación con Tricivir® comprimidos. Otros medicamentos, entre ellos, pero no limitados, los siguientes: aspirina, codeína, morfina, metadona, indometacina, ketoprofeno, naproxeno, oxazepam, lorazepam, cimetidina, clofibrato, dapsona e isoprinosina, pueden alterar el metabolismo de zidovudina al inhibir por competencia la glucuronidación o al inhibir directamente el metabolismo microsómico hepático. Las posibilidades de interacción entre estos medicamentos y Tricivir® comprimidos se deben analizar cuidadosamente antes de emplearlos, particularmente en terapia crónica. El tratamiento concomitante, en especial la terapia aguda, con medicamentos potencialmente nefrotóxicos o mielosupresores (por ejemplo, pentamidina, dapsona, pirimetamina, cotrimoxazol, amfotericina, flucitosina, ganciclovir, interferón, vincristina, vinblastina y doxorubicina sistémicos) también pueden aumentar el riesgo de reacciones adversas a zidovudina. Si se requiere de un tratamiento concomitante de Tricivir® comprimidos con cualquiera de estos medicamentos, deberá tenerse una precaución especial adicional, vigilando la función renal y los parámetros hematológicos y, en caso de que se requiera, debe reducirse la dosis de uno o más de los fármacos. Debido a que algunos pacientes que reciben Tricivir® comprimidos pueden seguir experimentando infecciones oportunistas, quizás deba considerarse el empleo concomitante de la terapia profiláctica antimicrobiana. Esta clase de profilaxis ha incluido cotrimoxazol, pentamidina en aerosol, pirimetamina y aciclovir. Los datos que se limitan a las pruebas clínicas no indican un aumento significativo en el riesgo de reacciones adversas a zidovudina con el uso de estos medicamentos.
|
|
Sobredosificación:
No se tiene experiencia en la sobredosificación con Tricivir® comprimidos. No se han identificado síntomas o signos específicos posteriores a una sobredosis aguda con zidovudina o lamivudina adicionales a los enumerados como efectos indeseables. No hubo fallecimientos y todos los pacientes se recuperaron. En los estudios clínicos, se han administrado a los pacientes dosis simples de hasta 1.200 mg y dosis diarias de hasta 1.800 mg de abacavir. No se reportaron reacciones adversas inesperadas. Se desconocen los efectos de dosis mayores. En caso de sobredosis, el paciente deben someterse a supervisión en búsqueda de evidencia de toxicidad (véase Efectos colaterales) y hay que aplicar el tratamiento ordinario de soporte, según se requiera. Ya que lamivudina puede eliminarse por diálisis, para el tratamiento de una sobredosis podría utilizarse la hemodiálisis, aunque ésta no se ha estudiado. La hemodiálisis y la diálisis peritoneal parecen tener un mínimo efecto sobre la eliminación de zidovudina; sin embargo, aumentan la eliminación del metabolito glucurónido. Se desconoce si abacavir puede eliminarse por diálisis peritoneal o por hemodiálisis. Para mayores detalles, el médico debe referirse a la información individual para prescribir de lamivudina, abacavir y zidovudina.
|
|
Incompatibilidades:
No aplicable.
|
|
Conservación:
Vida útil: La fecha de caducidad se indica en el empaque. Precauciones especiales de almacenamiento: No almacenar a una temperatura superior a 30şC.
|
|
Observaciones:
Importante: Tarjeta de alerta: Tricivir MR (sulfato de abacavir/lamivudina/zidovudina) en tabletas. Lleve esta tarjeta consigo en todo momento. Como Tricivir contiene abacavir, es posible que algunos pacientes que reciben tratamiento con Tricivir desarrollen alguna reacción de hipersensibilidad (reacción alérgica grave), la cual puede ser potencialmente mortal si se sigue administrando el tratamiento con Tricivir. Contecte a su médico inmediatamente para que le indique si debe suspender su tratamiento con Tricivir en caso de: 1) Experimentar algún exantema. 2) Experimentar uno o más síntomas de cuando menos 2 de los siguientes grupos: fiebre, disnea, dolor de garganta o tos, náuseas o vómito o diarrea o dolor abdominal, dolor o cansancio severo o malestar general. Si ha suspendido el tratamiento con Tricivir debido a esta reacción, nunca debe tomar nuevamente Tricivir ni cualquier otro medicamento que contenga abacavir (Kivexa, Ziagen), ya que en cuestión de horas podría experimentar un descenso potencialmente mortal en su presión arterial o incluso la muerte. Panel de la caja: Se debe incluir el siguiente texto en alguno de los paneles de la caja: Desprenda la Tarjeta de Alerta adjunta, ya que contiene información importante sobre seguridad. Advertencia: En caso de presentar cualquier síntoma que sugiera alguna reacción de hipersensibilidad, contacte a su médico inmediatamente. "Tire de aquí" (con la tarjeta de alerta adjunta).
|
|
Presentaciones:
Los comprimidos de Tricivir® se presentan en blister opaco blanco, o bien en frascos de polietileno de alta densidad con cerradura a prueba de nińos, conteniendo 60 comprimidos.
|
|



{kind=link}