 |
Composiciµn:
Cada ml de soluciµn para perfusiµn contiene: Cetuximab 5.000 mg; Cloruro de Sodio 5.844 mg; Glicina 7.507 mg; Polisorbato 80 0.100 mg; êcido CÚtrico Monohidrato 2.101 mg; Hidrµxido de Sodio 1 M c.s.p. pH = 5.5; Agua para Inyectables c.s.p. 1.00 ml.
|
|
Acciµn TerapÕutica:
Agente antineoplÃsico, anticuerpo monoclonal.
|
|
Indicaciones:
ErbituxÛ està indicado para el tratamiento de pacientes con cÃncer colorrectal metastÃsico con expresiµn del receptor del factor de crecimiento epidÕrmico (EGFR), con gen KRAS de tipo natural. En combinaciµn con quimioterapia basada en irinotecan o perfusiµn continua de 5-fluorouracilo/Ãcido folÚnico mÃs oxaliplatino (por detalles, ver secciµn Propiedades) en monoterapia en aquellos pacientes en los que haya fracasado el tratamiento con oxaliplatino e irinotecan y que no toleren Irinotecan. ErbituxÛ està indicado para el tratamiento de pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello en combinaciµn con radioterapia para la enfermedad localmente avanzada en combinaciµn con quimioterapia basada en el platino (cisplatino o carboplatino) para la enfermedad recurrente y/o metastÃsica. Como agente ºnico despuÕs del fracaso de la quimioterapia para la enfermedad recurrente y/o metastÃsica.
|
|
Propiedades:
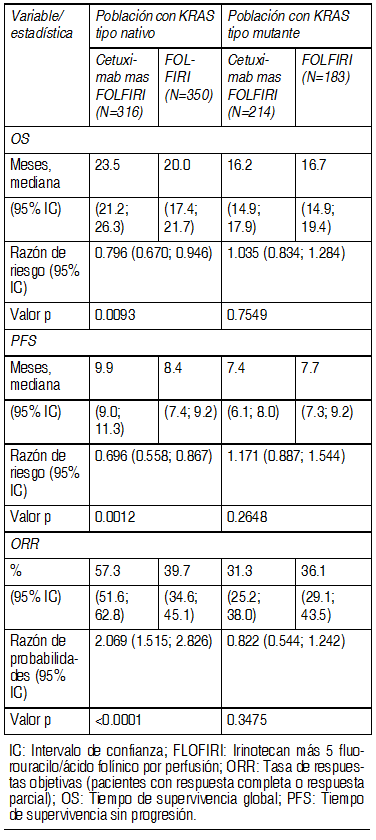
Mecanismo de acciµn: El Receptor del Factor de Crecimiento EpidÕrmico (EGFR) es parte de las vÚas de seþalizaciµn involucradas en el control de la sobrevida celular, progresiµn del ciclo celular, angiogÕnesis, migraciµn e invasiµn celular/metÃstasis. Cetuximab es un anticuerpo monoclonal IgG1 quimÕrico dirigido especÚficamente contra el EGFR. Se une a EGFR con una afinidad 5 a 10 veces mayor que los ligandos endµgenos y bloquea la funciµn del receptor. Induce la internalizaciµn de EGFR y de ese modo puede llevar a la regulaciµn negativa (down-regulation) de EGFR. Cetuximab tambiÕn hace que las cÕlulas efectoras citotµxicas del sistema inmune ataquen a las cÕlulas tumorales que expresan EGFR (citotoxicidad mediada por cÕlulas dependiente de anticuerpos, ADCC). Cetuximab no se une a otros receptores de la familia HER. El producto proteico del proto-oncogen KRAS (homµlogo del oncogen viral Kirsten 2 del sarcoma de rata) es un transductor central de seþales de EGFR ciclo abajo (downstream). En los tumores, la activaciµn de KRAS por parte de EGFR contribuye con un aumento de la proliferaciµn mediada por EGFR, la sobrevida y la producciµn de factores proangiogÕnicos. KRAS es uno de los oncogenes que se activan mÃs frecuentemente en los cÃnceres humanos. Las mutaciones del gen KRAS provocan la activaciµn constitutiva de la proteÚna KRAS independientemente de las seþales EGFR. Efectos farmacodinÃmicos: Cetuximab inhibe la proliferaciµn de cÕlulas tumorales humanas que expresan EGFR e induce la apoptosis. TambiÕn inhibe la producciµn de factores angiogÕnicos, bloquea la migraciµn de las cÕlulas endoteliales y produce una reducciµn de la neovascularizaciµn tumoral y de metÃstasis. CÃncer colorrectal: Se utilizµ un ensayo diagnµstico (EGFR pharmDxTM) para la detecciµn inmunohistoquÚmica de la expresiµn de EGFR en el material tumoral. Aproximadamente 75% de los pacientes con cÃncer colorrectal metastÃsico sometidos a tamizaje para estudios clÚnicos tenÚan un tumor que expresaba EGFR y por lo tanto eran considerados elegibles para el tratamiento con cetuximab. En el cÃncer colorrectal metastÃsico, la incidencia de mutaciones de KRAS està en el rango de 30 - 50%. Estudios recientes que muestran que los pacientes con cÃncer colorrectal metastÃsico con tumor KRAS de tipo natural tienen una posibilidad significativamente mayor de beneficiarse con el tratamiento con cetuximab o una combinaciµn de cetuximab y quimioterapia. Se investigµ el uso de cetuximab como agente ºnico o en combinaciµn con quimioterapia en 5 estudios clÚnicos controlados aleatorizados y varios estudios de respaldo. Los 5 estudios aleatorizados investigaron un total de 3734 pacientes con cÃncer colorrectal metastÃsico, con expresiµn detectable de EGFR y que tenÚan un estatus de desempeþo ECOG de È 2. La mayorÚa de los pacientes incluidos tenÚan un estatus de desempeþo ECOG de È 1. El estado KRAS fue reconocido como un factor predictivo para el tratamiento con cetuximab en 4 de los estudios controlados aleatorizados. Se contaba con el estado de mutaciµn KRAS para 2072 pacientes. El ºnico estudio en el que no se pudo hacer el anÃlisis fue el EMR 62 202-007. Cetuximab en combinaciµn con quimioterapia EMR 62 202-013: Este estudio aleatorizado en pacientes con cÃncer colorrectal metastÃsico que no habÚan recibido tratamiento previo para enfermedad metastÃsica comparµ la combinaciµn de cetuximab e irinotecan mÃs 5-Fluorouracilo/êcido folÚnico (FOLFIRI) por perfusiµn (599 pacientes) con la misma quimioterapia sola (599 pacientes). La proporciµn de pacientes con tumores KRAS de tipo natural fue el 63% de la poblaciµn de pacientes evaluable para el estado KRAS. Los datos de eficacia generados en este estudio se resumen en esta tabla:
Ver Tabla
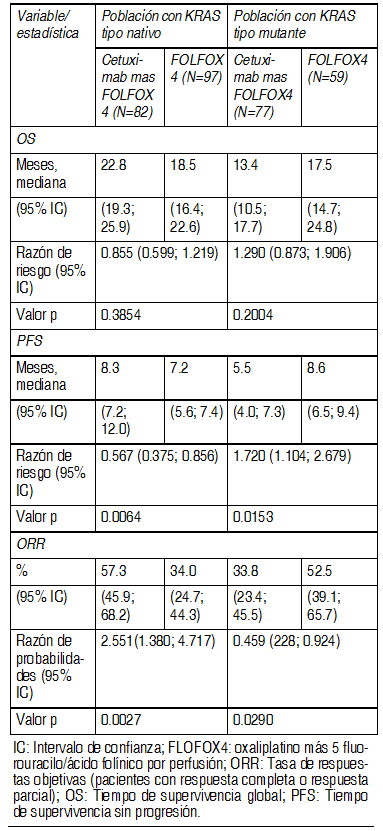
En la poblaciµn global el agregado de cetuximab a irinotecan mÃs 5-fluoruracilo/ acido folÚnico (FOLFIRI), mejorµ significativamente el tiempo de supervivencia global, tiempo de supervivencia libre de progresiµn y la tasa de respuestas objetivas. Los pacientes con tumores con KRAS no mutados y un estado de desempeþo ECOG de > 2 o de 65 aþos de edad o mayores, no se beneficiaron en tÕrminos de sobrevida total, al agregar cetuximab a FOLFIRI. EMR 62 202-047: Este estudio aleatorizado en pacientes con cÃncer colorrectal metastÃsico que no habÚan recibido tratamiento previo para enfermedad metastÃsica comparµ la combinaciµn de cetuximab y oxaliplatino mÃs 5-Fluorouracilo/êcido folÚnico por perfusiµn continua (FOLFOX4) (169 pacientes) con la misma quimioterapia sola (168 pacientes). La proporciµn de pacientes con tumores KRAS tipo natural fue 57% de la poblaciµn de pacientes evaluable para el estado KRAS. Los datos de eficacia generados en este estudio se resumen en la siguiente tabla:
Ver Tabla
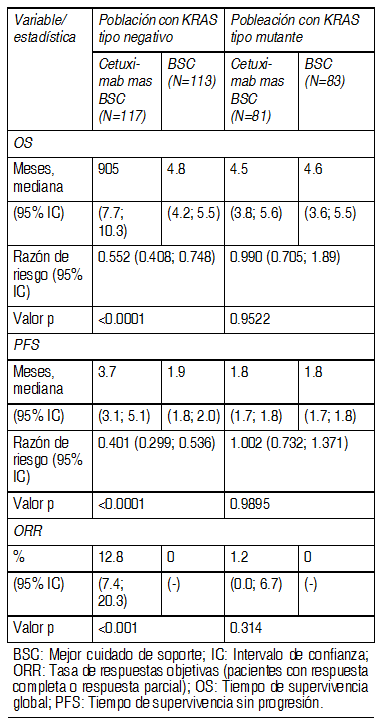
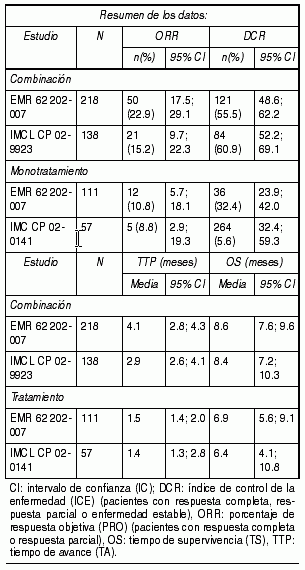
CA225006: Este estudio aleatorizado en pacientes con cÃncer colorrectal metastÃsico que habÚan recibido tratamiento de combinaciµn inicial con oxaliplatino mÃs fluoropirimidina para enfermedad metastÃsica comparµ la combinaciµn de cetuximab e irinotecan (648 pacientes) con irinotecan solo (650 pacientes). Al presentarse progresiµn de la enfermedad, se iniciµ tratamiento con agentes selectivos de EGFR en 50% de los pacientes en el brazo de irinotecan solo. En la poblaciµn general, independientemente del estado de KRAS, los resultados comunicados para cetuximab mÃs irinotecan (648 pacientes) contra irinotecan solo (650 pacientes) fueron los siguientes: mediana del tiempo de sobrevida total (OS) 10.71 vs. 9.99 meses (HR 0.98), mediana del tiempo de sobrevida libre de enfermedad (PFS) 4.0 vs. 2.6 meses (HR 0.69), y la tasa de respuesta objetiva (ORR) 16.4% vs. 4.2%. Con respecto al estado de KRAS, se contµ con sµlo 23% de los pacientes (300 de 1298). De la poblaciµn en la que se evaluµ KRAS, 64% de los pacientes (192) tenÚan tumores con KRAS no mutado y 108 pacientes con mutaciones de KRAS. Sobre la base de esta informaciµn, y dado que no se realizµ ninguna revisiµn independiente de los datos de imagenologÚa, se considera que estos resultados vinculados al estado de mutaciµn no son interpretables. EMR 62 202-007: Este estudio aleatorizado en pacientes con cÃncer colorrectal metastÃsico despuÕs de la falla del tratamiento basado en Irinotecan por enfermedad metastÃsica como ºltimo tratamiento antes de entrada al estudio comparµ la combinaciµn de cetuximab e irinotecan (218 pacientes) con la monoterapia con cetuximab (111 pacientes). La combinaciµn de cetuximab con irinotecan comparado con cetuximab solo, redujo el riesgo global de progresiµn de la enfermedad en 46% y aumentµ significativamente la tasa de respuesta objetiva. En el estudio aleatorizado, la mejorÚa del tiempo de sobrevida global no alcanzµ significaciµn estadÚstica; sin embargo, en el tratamiento de seguimiento, casi 50% de los pacientes del brazo que recibiµ solo cetuximab recibiµ una combinaciµn de cetuximab e irinotecan despuÕs de la progresiµn de la enfermedad, lo que puede haber influido en el tiempo de sobrevida global. Cetuximab como ºnico agente: CA225025: Este estudio aleatorizado en pacientes con cÃncer colorrectal metastÃsico que habÚan recibido tratamiento previo basado en oxaliplatino, irinotecan y fluoropirimidina por enfermedad metastÃsica comparµ el agregado de cetuximab como agente ºnico para el mejor cuidado de soporte (BSC) (287 pacientes) con mejor cuidado de soporte (285 pacientes). La proporciµn de pacientes con tumores KRAS tipo natural fue 58% de la poblaciµn de pacientes evaluables para KRAS. Los datos de eficacia obtenidos en este ensayo se resumen en la siguiente tabla:
Ver Tabla
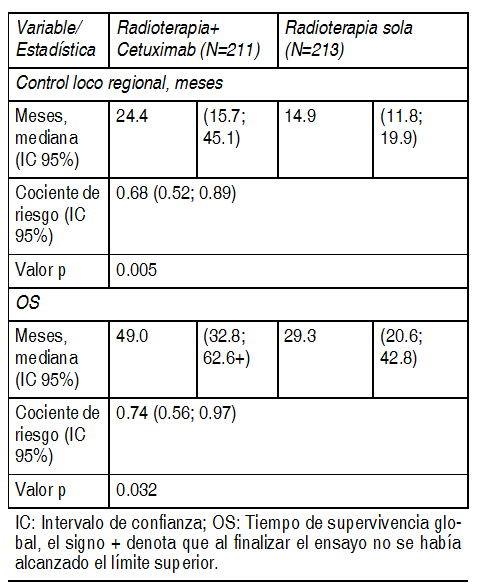
CÃncer de cÕlulas escamosas de cabeza y cuello: La detecciµn inmunohistoquÚmica de la expresiµn de EGFR no se realizµ, dado que mÃs del 90% de los pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello tienen tumores que expresan EGFR. Cetuximab en combinaciµn con radioterapia para enfermedad localmente avanzada: EMR 62 202-006: Este estudio aleatorizado comparµ la combinaciµn de cetuximab y radioterapia (211 pacientes) con radioterapia sola (213 pacientes) en pacientes con cÃncer de cÕlulas escamosas localmente avanzado de cabeza y cuello. Se iniciµ cetuximab 1 semana antes de la radioterapia y se administrµ hasta el final del perÚodo de radioterapia. Los datos de eficacia generados en este estudio se resumen en la siguiente tabla:
Ver Tabla
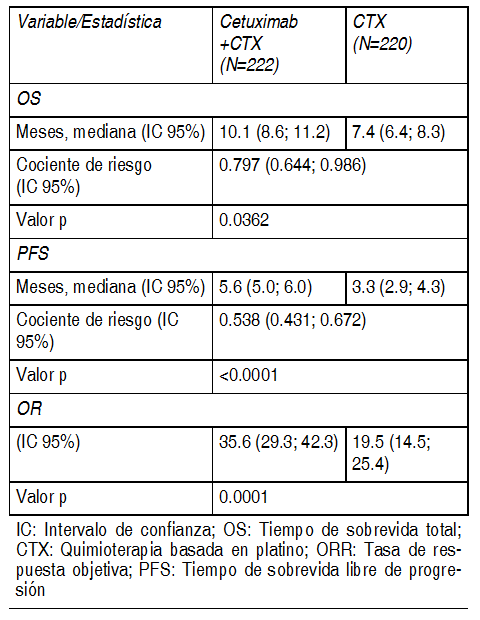
Los pacientes con buen pronµstico, indicado por el estadio del tumor, estado de desempeþo de Karnofsky (KPS) y edad tuvieron un beneficio mÃs pronunciado al agregar cetuximab a la radioterapia. No se pudo demostrar beneficio clÚnico en pacientes con KPS È 80 que tuvieran 65 aþos de edad o mÃs. El uso de cetuximab en combinaciµn con la quimio-radioterapia hasta ahora no se ha investigado adecuadamente. Por lo tanto, todavÚa no se ha logrado establecer una relaciµn riesgo-beneficio para esta combinaciµn. Cetuximab en combinaciµn con quimioterapia basada en platino y enfermedad recurrente y/o metastÃsica: EMR 62 202-002: Este estudio aleatorizado en pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello recurrente y/o metastÃsico que no hubieran recibido antes quimioterapia para esta enfermedad comparµ la combinaciµn de cetuximab y cisplatino o carboplatino mÃs 5-fluorouracilo por perfusiµn (222 pacientes) con la misma quimioterapia sola (220 pacientes). El tratamiento del brazo con cetuximab consistiµ en hasta 6 cursos de quimioterapia basada en platino en combinaciµn con cetuximab seguidos de cetuximab como terapia de mantenimiento hasta la progresiµn de la enfermedad. Los datos de eficacia generados por este estudio se resumen en la siguiente tabla:
Ver Tabla
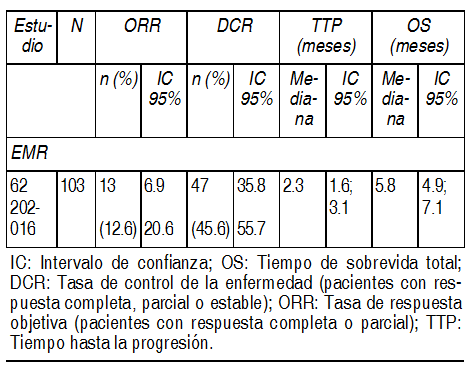
Los pacientes con buen pronµstico, indicado por el estadio del tumor, el estado de funcionamiento de Karnofsky (KPS) y la edad tuvieron un beneficio mÃs pronunciado al agregar cetuximab a la quimioterapia en base a platino. A diferencia del tiempo de sobrevida libre de progresiµn, no se pudo demostrar ningºn beneficio en el tiempo de sobrevida total en los pacientes con KPS È 80 de 65 aþos o mayores. Cetuximab como agente ºnico luego del fracaso de la quimioterapia para enfermedad recurrente y/o metastÃsica: EMR 62 202-016: Este estudio abierto con ºnico brazo investigµ el tratamiento con cetuximab como ºnico agente en 103 pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello recurrente o metastÃsico luego del fracaso de la quimioterapia de primera lÚnea. Los datos de eficacia generados en este estudio se resumen en la siguiente tabla:
Ver Tabla
Inmunogenicidad: El desarrollo de anticuerpos antiquimÕricos humanos (HACA) es un efecto especÚfico de clase de los anticuerpos quimÕricos monoclonales. Aparecieron tÚtulos mensurables de HACA en 3.4% de los pacientes estudiados. No se dispone a la fecha de ningºn dato concluyente sobre el efecto neutralizante sobre cetuximab. La apariciµn de HACA no se correlacionµ con la presentaciµn de reacciones de hipersensibilidad o ningºn otro efecto indeseado de cetuximab. Propiedades farmacocinÕticas: Las infusiones intravenosas de cetuximab presentaron farmacocinÕtica dosis-dependiente en dosis semanales desde 5 a 500 mg/m2 de Ãrea superficie corporal. Cuando se administrµ cetuximab a una dosis inicial de 400 mg/m2 Ãrea superficie corporal, el volumen de distribuciµn medio fue aproximadamente equivalente al espacio vascular (2.9 l/m2 con un rango de 1.5 a 6.2 l/m2). La Cmax media (Ý desviaciµn estÃndar) fue 185Ý55 microgramos por ml. La depuraciµn media fue 0.022 l/h por m2 de Ãrea superficie corporal. Cetuximab tiene una vida media de eliminaciµn prolongada, con valores que van de 70 a 100 horas a la dosis objetivo. Las concentraciones sÕricas de cetuximab alcanzaron niveles estables despuÕs de 3 semanas de monoterapia con cetuximab. Las concentraciones pico medias de cetuximab fueron 155.8 mcg por ml en la semana 3 y 151.6 mcg por ml en la semana 8, mientras que las concentraciones valle medias correspondientes fueron 41.3 y 55.4 mcg por ml, respectivamente. En un estudio de cetuximab administrado en combinaciµn con IrinotecÃn, los niveles valle medios de cetuximab fueron 50.0 mcg por ml en la semana 12 y 49.4 mcg por ml en la semana 36. Se han descrito varias vÚas que pueden contribuir al metabolismo de los anticuerpos. En todas estas vÚas interviene la biodegradaciµn del anticuerpo a molÕculas mÃs pequeþas. Un anÃlisis integrado de todos los estudios clÚnicos mostrµ que las caracterÚsticas farmacocinÕticas de cetuximab no se ven influidas por raza, edad, gÕnero y funcionalidad renal o hepÃtica. En un estudio Fase I en pacientes pediÃtricos (1-18 aþos) con tumores sµlidos refractarios, se administrµ cetuximab en combinaciµn con irinotecan. Los resultados farmacocinÕticos fueron comparables a los de adultos. Datos de seguridad no clÚnicos: Los principales hallazgos de los estudios de toxicidad en animales fueron las alteraciones cutÃneas dependientes de las dosis, comenzando a niveles de dosis equivalentes a los usados en humanos. Un estudio de toxicidad embriofetal en los monos Cynomolgus no revelµ signos de teratogenicidad. Sin embargo, dependiendo de la dosis, se observµ un aumento de la incidencia de abortos. Los datos no clÚnicos sobre la genotoxicidad y la tolerancia local, incluyendo vÚas de administraciµn accidentales, revelaron que no habÚa peligros especiales para humanos. No se han realizado estudios formales en animales para establecer el potencial carcinogÕnico de cetuximab o para determinar sus efectos sobre la fertilidad masculina y femenina. No se han realizado estudios de toxicidad con la coadministraciµn de cetuximab y agentes quimioterÃpicos. No se dispone a la fecha de datos no clÚnicos sobre el efecto de cetuximab en la cicatrizaciµn de heridas. Sin embargo, en los modelos preclÚnicos de curaciµn de heridas, los inhibidores de tirosina quinasa selectivos de EGFR demostraron retrasar la cicatrizaciµn de las heridas.
|
|
PosologÚa:
ErbituxÛ debe ser administrado bajo la supervisiµn de un mÕdico experimentado en el uso de los productos medicinales antineoplÃsicos. Se necesita un monitoreo cercano durante el tiempo de perfusiµn y por lo menos 1 hora despuÕs de finalizar la misma. Debe disponerse de equipos de reanimaciµn. Antes de la primera perfusiµn, los pacientes deben recibir premedicaciµn con un antihistamÚnico y un corticosteroide. Esta premedicaciµn se recomienda antes de todas las infusiones subsiguientes. En todas las indicaciones, se administrµ ErbituxÛ 1 vez por semana. La primera dosis es 400 mg de ErbituxÛ por m2 de Ãrea de superficie corporal con un perÚodo de perfusiµn recomendado de 120 minutos. Todas las dosis semanales subsiguientes son 250 mg por m2 de Ãrea de superficie corporal, cada uno con un perÚodo de perfusiµn recomendado de 60 minutos. La velocidad de perfusiµn mÃxima no debe superar los 10 mg/min. CÃncer colorrectal: En pacientes con cÃncer colorrectal metastÃsico, se utiliza ErbituxÛ en combinaciµn con quimioterapia o como agente ºnico. Se recomienda realizar la detecciµn del estado mutacional de KRAS antes de la primera perfusiµn de Erbitux. Es importante que un mÕtodo de ensayo validado es utilizado por un laboratorio experimentado. Para la dosificaciµn o para las modificaciones de dosis recomendadas de los agentes quimioterÃpicos usados concomitantemente, por favor referirse a la informaciµn para prescribir de estos productos medicinales. No deben administrarse hasta tanto no haya transcurrido 1 hora de la finalizaciµn de la perfusiµn con ErbituxÛ. Se recomienda continuar con el tratamiento con ErbituxÛ hasta la progresiµn la enfermedad. CÃncer de cÕlulas escamosas de cabeza y cuello: En los pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello localmente avanzado, se utiliza ErbituxÛ concomitantemente con radioterapia. Se recomienda iniciar la terapia con ErbituxÛ 1 semana antes de la radioterapia y continuar el tratamiento con ErbituxÛ hasta el final del perÚodo de radioterapia. En pacientes con cÃncer de cÕlulas escamosas de cabeza y cuello recurrente y/o metastÃsico, se utiliza ErbituxÛ en combinaciµn con quimioterapia basada en platino, seguido de ErbituxÛ como terapia de mantenimiento hasta la progresiµn de la enfermedad. No debe administrarse quimioterapia hasta tanto no haya transcurrido 1 hora de la finalizaciµn de la perfusiµn con ErbituxÛ. En los pacientes en los que fracasµ la quimioterapia para cÃncer de cÕlulas escamosas de cabeza y cuello recurrente y/o metastÃsico, tambiÕn se usa cetuximab como agente ºnico. Se recomienda continuar con el tratamiento con ErbituxÛ hasta la progresiµn de la enfermedad.
|
|
Modo de Empleo:
Instrucciones para uso y manejo: Se administra ErbituxÛ 5 mg/ml por vÚa intravenosa con una bomba de perfusiµn, goteo por gravedad o una bomba con jeringa. Debe utilizarse una vÚa de perfusiµn aparte para la perfusiµn, y debe enjuagarse la vÚa con una soluciµn para inyecciµn de cloruro de sodio estÕril 9 mg/ml (0.9%) al final de la perfusiµn. ErbituxÛ 5 mg/ml es una soluciµn incolora. ErbituxÛ 5 mg/ml es compatible con: Bolsas de PE (polietileno), EVA (etil vinil acetato) o PVC (polivinil cloruro), aparatos de perfusiµn de PE, EVA, PVC, TP (poliolefino termoplast) o PUR (poliuretano), jeringas para bombas de jeringas de PP (polipropileno). ErbituxÛ no contiene conservantes antimicrobianos o agentes bacteriostÃticos. Por lo tanto, al preparar la perfusiµn debe asegurarse un manejo asÕptico. Se recomienda enfÃticamente usar inmediatamente despuÕs de abrir. ErbituxÛ 5 mg/ml debe prepararse de la siguiente manera: Para administraciµn con bomba de perfusiµn o goteo por gravedad (diluido con soluciµn de cloruro de sodio estÕril de 9 mg/ml [0.9%]): Tomar una bolsa de perfusiµn de un tamaþo adecuado de soluciµn de cloruro de sodio estÕril de 9 mg/ml (0.9%). Calcular el volumen necesario de ErbituxÛ. Extraer un volumen adecuado de la soluciµn de cloruro de sodio de la bolsa de perfusiµn, utilizando una jeringa estÕril apropiada con una aguja adecuada. Tomar una jeringa estÕril apropiada y adosar una aguja adecuada. Extraer el volumen necesario de ErbituxÛ de un vial. Transferir ErbituxÛ en la bolsa de perfusiµn preparada. Repetir este procedimiento hasta alcanzar el volumen calculado. Conectar la vÚa de perfusiµn y cebarla con el ErbituxÛ diluido antes de iniciar la perfusiµn. Utilizar un goteo por gravedad o una bomba de perfusiµn para administraciµn. Programar y controlar la velocidad como se explica arriba. Para administraciµn con bomba de perfusiµn o goteo por gravedad (no diluido): Calcular el volumen necesario de ErbituxÛ. Tomar una jeringa estÕril apropiada (mÚnimo 50 ml) y adosar una aguja adecuada. Extraer el volumen adecuado de ErbituxÛ de un vial. Transferir el ErbituxÛ a un recipiente evacuado estÕril o bolsa. Repetir este procedimiento hasta alcanzar el volumen calculado. Conectar la vÚa de perfusiµn y cebarla con ErbituxÛ antes de comenzar la perfusiµn. Utilizar un goteo por gravedad o una bomba de perfusiµn para administraciµn. Configurar y controlar la velocidad como se explica antes. Para administraciµn con una bomba de jeringa: Calcular el volumen requerido de ErbituxÛ. Tomar una jeringa estÕril apropiada y adosar una aguja adecuada. Extraer el volumen necesario de ErbituxÛ de un vial. Retirar la aguja y poner la jeringa en la bomba de la jeringa. Conectar la vÚa de perfusiµn a la jeringa, configurar y controlar la velocidad tal como se explica antes y comenzar la perfusiµn despuÕs de cebar la vÚa con ErbituxÛ o soluciµn de cloruro de sodio estÕril 9 mg/ml (0.9%). De ser necesario, repetir este procedimiento hasta infundir el volumen calculado.
|
|
Efectos Colaterales:
Los efectos adversos mÃs adelante enumerados pueden esperarse bajo tratamiento con ErbituxÛ. Las siguientes definiciones se aplican a la terminologÚa de frecuencia utilizada de aquÚ en adelante: Muy comºn ( ° 1/10); Comºn ( ° 1/100 a < 1/10); No comºn ( ° 1/1000 a < 1/100); Raro ( ° 1/10000 a < 1/1000); Muy raro (< 1/10000); Frecuencia no conocida (no se puede estimar a partir de los datos disponibles); Un asterisco (*) indica que se presenta informaciµn adicional sobre el efecto indeseado respectivo abajo de la tabla. Trastornos del sistema nervioso: Comºn: Cefaleas. Frecuencia no conocida: Meningitis asÕptica. Trastornos oculares: Comºn: Conjuntivitis. No comºn: Blefaritis, queratitis. Trastornos respiratorios, torÃcicos y mediastinales: No comºn: Embolismo pulmonar. Raro: Enfermedad pulmonar intersticial. Trastornos gastrointestinales: Comºn: Diarrea, nÃuseas, vµmitos. Trastornos cutÃneos y de tejidos subcutÃneos: Muy comºn: Reacciones cutÃneas*. Frecuencia no conocida: Sobreinfecciµn de lesiones cutÃneas *. Muy raro: SÚndrome de Stevens-Johnson, necrosis epidÕrmica tµxica. Trastornos del metabolismo y la nutriciµn: Muy comunes: Hipomagnesemia. Comunes: Deshidrataciµn, en particular secundaria a diarrea o mucositis; hipocalcemia; anorexia que puede provocar adelgazamiento. Trastornos vasculares: No comºn: Trombosis venosa profunda. Trastornos generales y condiciones del sitio de administraciµn: Muy comunes: Reacciones leves o moderadas relacionadas con la perfusiµn*; mucositis en algunos casos severa. La mucositis puede llevar a epistasis. Comunes: Reacciones severas relacionadas con la perfusiµn*, fatiga. Trastornos hepatobiliares: Muy comunes: Aumento de los niveles de las enzimas hepÃticas (ASAT, ALAT, AP). Informaciµn adicional: En tÕrminos generales, no se observµ ninguna diferencia clÚnica relevante entre gÕneros. Reacciones vinculadas a la perfusiµn: Es muy comºn ver reacciones a la perfusiµn leve o moderadas, comprendiendo sÚntomas como fiebre, escalofrÚos, mareos, o disnea, los que ocurren cercanos en el tiempo, fundamentalmente con la primera perfusiµn de ErbituxÛ. Es posible que comºnmente aparezcan reacciones severas relacionadas con la perfusiµn, en raros casos con desenlace fatal. Habitualmente aparecen durante o dentro de la primera hora de la perfusiµn inicial de ErbituxÛ, pero pueden aparecer tras varias horas o con las infusiones subsiguientes. Si bien aºn no se han identificado los mecanismos subyacentes, algunas de estas reacciones pueden ser de naturaleza anafilactoide/anafilÃctica y pueden incluir sÚntomas como broncoespasmo, urticaria, aumento o disminuciµn de la presiµn sanguÚnea, pÕrdida de conciencia o shock. En raros casos se ha descrito la apariciµn de angina de pecho, infarto miocÃrdico o paro cardÚaco. Para el manejo clÚnico de las reacciones relacionadas con la perfusiµn, ver "Advertencias y Precauciones especiales". Reacciones cutÃneas: Pueden aparecer reacciones cutÃneas en mÃs de 80% de los pacientes, y se presentan fundamentalmente como una erupciµn tipo acnÕ y/o, con menor frecuencia, como prurito, piel seca, descamaciµn, hipertricosis, o trastornos ungueales (por ejemplo: paroniquia). Aproximadamente 15% de las reacciones cutÃneas son severas, incluyendo casos ºnicos de necrosis cutÃnea. La mayorÚa de las reacciones cutÃneas se presentan dentro de las 3 primeras semanas de tratamiento. Generalmente se resuelven sin secuelas, con el tiempo, despuÕs de interrumpir el tratamiento, si se siguen los ajustes recomendados en el rÕgimen posolµgico. Las lesiones cutÃneas inducidas por ErbituxÛ pueden predisponer a los pacientes a sobreinfecciones (por ejemplo con S. aureus), lo que puede llevar a complicaciones posteriores, como celulitis, erisipelas, o, potencialmente, puede haber un desenlace fatal por sÚndrome de piel escaldada estafilocµcico, fascitis necrotizante o sepsis. Tratamiento combinado: Cuando se utilice ErbituxÛ en combinaciµn con agentes quimioterÃpicos, referirse tambiÕn a la informaciµn para prescribir de estos productos medicinales. Por efectos adversos en combinaciµn con otros agentes quimioterÃpicos, referirse a "Interacciones". En combinaciµn con radioterapia local de cabeza y cuello, los otros efectos indeseables fueron los habituales de la radioterapia (como mucositis, dermatitis rÃdica, disfagia o leucopenia, presentÃndose fundamentalmente como linfocitopenia). En un estudio clÚnico controlado, aleatorizado, con 424 pacientes, que declaraban tasas de dermatitis rÃdica severa aguda y mucositis, asÚ como eventos tardÚos relacionados con la radioterapia, fueron levemente mÃs altos en pacientes que recibÚan radioterapia en combinaciµn con ErbituxÛ, que en los que recibÚan radioterapia sola.
|
|
Contraindicaciones:
Erbitux està contraindicado en pacientes con reacciones de hipersensibilidad severas (grados 3 µ 4; Instituto Nacional de CÃncer de E.U.A - Criterios Comunes de TerminologÚa para Eventos Adversos; CTCAE) provocadas por cetuximab. Antes de iniciar el tratamiento combinado, deben considerarse las contraindicaciones para el uso concomitante de agentes quimioterÃpicos o radioterapia.
|
|
Advertencias:
Advertencias y precauciones especiales: Reacciones relacionadas con la perfusiµn: Si los pacientes presentan reacciones relacionadas con la perfusiµn leve o moderadas, puede reducirse la velocidad de perfusiµn. Se recomienda mantener una velocidad de perfusiµn menor en todas las infusiones subsiguientes. Se han comunicado reacciones severas relacionadas con la perfusiµn en pacientes tratados con ErbituxÛ. Los sÚntomas habitualmente se presentaron durante la primera perfusiµn y hasta 1 hora despuÕs de la finalizaciµn de la perfusiµn, pero puede aparecer varias horas despuÕs o con infusiones posteriores. Se recomienda advertir a los pacientes de la posibilidad de una apariciµn tan tardÚa y darles instrucciones de consultar a su mÕdico si se presentaran sÚntomas de reacciones relacionadas con la perfusiµn. Si hubiera reacciones severas vinculadas a la perfusiµn, se debe interrumpir el tratamiento con ErbituxÛ de manera inmediata y permanente y puede ser necesario instaurar un tratamiento de emergencia. Se recomienda tener atenciµn especial con los pacientes con estado funcional reducido y patologÚa cardiopulmonar preexistente. Trastornos respiratorios: Se han descrito casos de enfermedad pulmonar intersticial, perteneciendo la mayorÚa de los pacientes a la poblaciµn japonesa. Si se diagnostica enfermedad pulmonar intersticial, debe interrumpirse la administraciµn de Erbitux y el paciente debe ser tratado apropiadamente. Reacciones cutÃneas: Las reacciones cutÃneas son muy comunes y tal vez requieran interrumpir o suspender el tratamiento. Las directrices de prÃctica clÚnica indican que se debe considerar el uso de tetraciclinas por vÚa oral (6-8 semanas) y la aplicaciµn tµpica de crema de hidrocortisona al 1% con humectante. Se han utilizado corticoides tµpicos de alta potencia o tetraciclinas por vÚa oral para el tratamiento de las reacciones cutÃneas. Si un paciente presenta reacciones cutÃneas severas ( ° grado 3; Instituto Nacional del CÃncer de EUA - Criterios Comunes de Toxicidad para eventos adversos; CTCAE), debe interrumpirse el tratamiento con ErbituxÛ. Sµlo se puede retomar el tratamiento si la reacciµn mejora a un grado 2. Si es la primera vez que apareciµ una reacciµn cutÃnea severa, debe reinstaurarse el tratamiento sin cambiar las dosis. Al aparecer reacciones cutÃneas severas por segunda o tercera vez, debe interrumpirse nuevamente el tratamiento con ErbituxÛ. Sµlo se puede retomar el tratamiento a un nivel de dosis mÃs bajo (200 mg/m2 del Ãrea de superficie corporal despuÕs de la segunda apariciµn, y 150 mg/m2 despuÕs de la tercera apariciµn), si se ha resuelto la reacciµn al grado 2. Si aparecieran reacciones cutÃneas severas una cuarta vez o si no se resuelven a grado 2 durante la interrupciµn del tratamiento, debe interrumpirse el tratamiento con ErbituxÛ de manera permanente. Trastornos electrolÚticos: Es frecuente que se produzca una disminuciµn progresiva de los niveles sÕricos de magnesio, lo que puede llevar a una hipomagnesemia severa. La hipomagnesemia es reversible luego de interrumpir ErbituxÛ. AdemÃs, puede aparecer hipokalemia como consecuencia de diarrea. TambiÕn puede aparecer hipocalcemia; puede aumentar la frecuencia de hipocalcemia severa en particular en combinaciµn con una quimioterapia basada en platino. Se recomienda hacer la determinaciµn de los niveles sÕricos de los electrolitos antes de iniciar el tratamiento con ErbituxÛ y durante el mismo y se recomienda restituir los electrolitos, segºn corresponda. Neutropenia y complicaciones infecciosas relacionadas: Los pacientes que reciben cetuximab en combinaciµn con quimioterapia basada en platino tienen un mayor riesgo de apariciµn de neutropenia severa, lo que puede llevar a complicaciones infecciosas subsiguientes como neutropenia febril, neumonÚa o sepsis. En este tipo de pacientes se recomienda un monitoreo cuidadoso, en particular en aquellos que presentan lesiones cutÃneas, mucositis o diarrea que pueden facilitar la apariciµn de infecciones. Trastornos cardiovasculares: Se ha observado una frecuencia aumentada de acontecimientos cardiovasculares graves y a veces mortales y de muertes surgidas a raÚz del tratamiento en la terapia para el cÃncer de pulmµn no microcÚtico, el carcinoma de cÕlulas escamosas de cabeza y cuello y el carcinoma colorrectal. En algunos estudios, se ha observado una asociaciµn con una edad ° 65 aþos. Cuando se prescriba Erbitux, debe tenerse en cuenta el estado y desempeþo cardiovascular de los pacientes y la administraciµn concomitante de compuestos cardiotµxicos como las fluoropirimidinas. Trastornos oculares: Se han descrito casos de queratitis y queratitis ulcerosa con el uso de cetuximab. Se recomienda que los pacientes con signos y sÚntomas sugestivos de queratitis consulten a un oftalmµlogo. Si se diagnosticara queratitis, deben considerarse cuidadosamente los beneficios y los riesgos de continuar con el tratamiento. Si se confirma el diagnµstico de queratitis ulcerosa, debe interrumpirse o suspenderse el tratamiento con cetuximab. Se recomienda prestar atenciµn especial en los pacientes con antecedentes de queratitis, queratitis ulcerosa o con casos severos de ojo seco. Pacientes con cÃncer colorrectal cuyos tumores presentan mutaciones del gen KRAS: Erbitux no debe usarse para el tratamiento de los pacientes con cÃncer colorrectal cuyos tumores presenten mutaciones del gen KRAS o en los que se desconozca el estado tumoral con respecto a dicho gen. Los resultados de los estudios clÚnicos muestran un equilibrio riesgo-beneficio negativo en los tumores con mutaciones del gen KRAS en particular en combinaciµn con la perfusiµn continua de 5-fluorouracilo/ Ãcido folÚnico mÃs oxaliplatino (ver la Secciµn de Propiedades). Tratamiento combinado: Cuando se utilice ErbituxÛ en combinaciµn con agentes quimioterÃpicos, referirse tambiÕn a la informaciµn para prescribir de estos productos medicinales. Existe experiencia limitada en el uso de cetuximab en combinaciµn con la radioterapia en el cÃncer colorrectal.
|
|
Precauciones:
Embarazo y lactancia: El EGFR interviene en el desarrollo del feto. Observaciones limitadas en animales indican que habrÚa pasaje de cetuximab por la placenta, y se ha encontrado que otros anticuerpos IgG1 atraviesan la barrera placentaria. Los datos en animales no revelaron ninguna evidencia de teratogenicidad. Sin embargo, dependiendo de la dosis, se ha observado un aumento de la incidencia de abortos. No se dispone de suficiente informaciµn de mujeres embarazadas. Se recomienda firmemente administrar ErbituxÛ durante el embarazo o a cualquier mujer que no estÕ usando anticoncepciµn adecuada sµlo si el beneficio potencial justifica un riesgo potencial para el feto. Se recomienda que las mujeres no amamanten durante el tratamiento con ErbituxÛ y hasta 2 meses despuÕs de la ºltima dosis, porque no se sabe si ErbituxÛ se excreta en la leche de pecho. No hay datos sobre el efecto de cetuximab sobre la fertilidad humana. Los efectos sobre la fertilidad en el hombre y la mujer no han sido evaluados con estudios formales en animales. Efectos sobre la capacidad de conducir y usar mÃquinas: No se han realizado estudios sobre los efectos sobre la capacidad de conducir y usar mÃquinas. Si los pacientes presentan sÚntomas relacionados con el tratamiento que afecten su capacidad para concentrarse y reaccionar, se recomienda que no conduzcan o usen mÃquinas hasta que remitan los efectos. Poblaciones especiales: A la fecha sµlo se han investigado pacientes con una funciµn renal y hepÃtica adecuada (creatininemia È 1.5 veces, transaminasas È 5 veces y bilirrubina È 1.5 veces el lÚmite superior normal). No se ha estudiado ErbituxÛ en pacientes con uno o mÃs de los siguientes parÃmetros de laboratorio anormales: hemoglobina < 9 g/dl, conteo leucocitario < 3.000/mm3, conteo absoluto de neutrµfilos < 1.500/mm3, recuento plaquetario < 100.000/mm3. No se han establecido la seguridad ni la efectividad de ErbituxÛ en los pacientes pediÃtricos menores de 18 aþos. No se identificaron nuevas seþales de seguridad en pacientes pediÃtricos, tal como se comunicara en un estudio fase I. No se requieren ajustes de dosis en los ancianos, pero la experiencia con los pacientes de 75 aþos o mÃs es limitada.
|
|
Interacciones Medicamentosas:
En combinaciµn con la quimioterapia basada en platino, puede aumentar la frecuencia de leucopenia severa o neutropenia severa, y asÚ puede llevar a una tasa mÃs alta de complicaciones infecciosas como una neutropenia febril, neumonÚa y sepsis comparado con la quimioterapia basada solo en platino (ver tambiÕn "Advertencias especiales y precauciones"). En combinaciµn con fluoropirimidinas por perfusiµn, se vio un aumento de la frecuencia de isquemia cardÚaca, incluyendo el infarto de miocardio, la insuficiencia cardÚaca congestiva asÚ como la frecuencia del sÚndrome mano-pie (eritro disestesia palmo-plantar), comparado con la frecuencia observada con fluoropirimidinas. Un estudio de interacciµn formal con irinotecan en humanos mostrµ una farmacocinÕtica inalterada de ambas drogas al usarse en combinaciµn. Los datos clÚnicos no mostraron influencia sobre el perfil de seguridad de ErbituxÛ. No se han realizado otros estudios de interacciµn formales con ErbituxÛ en humanos. En combinaciµn con capecitabina y oxaliplatino (XELOX), se puede aumentar la frecuencia de diarrea severa.
|
|
Sobredosificaciµn:
Hay experiencia limitada con dosis ºnicas mayores a 400 mg/m2 de Ãrea de superficie corporal a la fecha o administraciones semanales de dosis mayores a 250 mg/m2 de Ãrea de superficie corporal.
|
|
Incompatibilidades:
No debe mezclarse ErbituxÛ 5 mg/ml con otros productos medicinales aplicados por vÚa intravenosa, excepto los mencionados arriba en la secciµn "Instrucciones para uso y manejo". Debe utilizarse una vÚa de perfusiµn aparte.
|
|
Conservaciµn:
Conservar en el refrigerador o heladera entre 2¤C y 8¤C. No utilizar despuÕs de la fecha de vencimiento. ErbituxÛ no contiene ningºn conservante antimicrobiano o agente bacteriostÃtico. Utilizar inmediatamente despuÕs de abrir. Una vez abierto, utilÚcelo inmediatamente, los tiempos de conservaciµn en uso y las condiciones son responsabilidad del usuario.
|
|
Observaciones:
Advertencia de este y todos los medicamentos: Mantenga todos los medicamentos fuera del alcance de los niþos. No utilizar despuÕs de la fecha de vencimiento. Ösese solo por indicaciµn y bajo supervisiµn mÕdica. No repita el medicamento sin indicaciµn del mÕdico. No utilice este medicamento si observa signos visibles de deterioro.
Ver Tabla
|
|
Presentaciones:
Envases conteniendo 1 vial de 20 ml.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}