 |
Composición:
Cada comprimido recubierto contiene: Abacavir 600 mg; Lamivudina 300 mg. Excipientes: Núcleo: Estearato de Magnesio, Celulosa Microcristalina, Almidón Glicolato de Sodio. Recubrimiento: Color Anaranjado Opadry YS-1-13065-A que contiene: Hipromelosa (E464), Dióxido de Titanio (E171), Macrogol 400, Polisorbato 80 (E433), Colorante FD&C Amarillo Nş6, Laca Alumínica. Forma farmacéutica: Comprimidos capsuliformes modificados, con recubrimiento pelicular, de color anaranjado, grabados en bajorrelieve con GS FC2 en un lado.
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: inhibidor nucleosídico de la transcriptasa inversa (INTI). Código ATC: J05A R02.
|
|
Indicaciones:
Kivexa es una combinación de dos análogos nucleosídicos (abacavir y lamivudina). Está indicada en la terapia antirretroviral combinada para el tratamiento de la infección causada por el Virus de la Inmunodeficiencia Humana (VIH) en adultos y adolescentes a partir de 12 ańos de edad.
|
|
Propiedades:
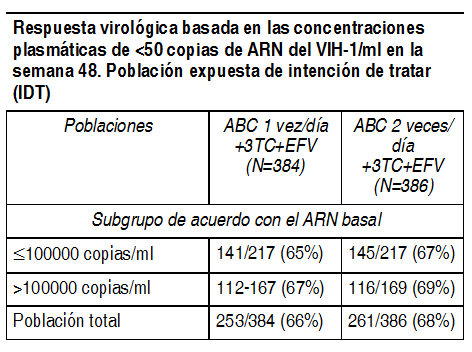
Propiedades farmacológicas: Farmacodinamia: Mecanismo de acción: El abacavir y la lamivudina son INTIs, y son inhibidores potentes y selectivos del virus de la inmunodeficiencia humana del tipo 1 (VIH 1) y del tipo 2 (VIH 2). Tanto el abacavir como la lamivudina son metabolizados secuencialmente por las cinasas intracelulares a los trifosfatos respectivos (TFs) que son las partes activas. El TF de lamivudina y el TF de carbovir (la forma trifosfato activa del abacavir) son sustratos e inhibidores competitivos de la transcriptasa inversa (TI) del virus de la inmunodeficiencia humana (VIH). Sin embargo, su principal actividad antiviral la ejerce mediante la incorporación de la forma monofosfato en la cadena del ácido desoxirribonucleico (ADN) viral, lo cual resulta en terminación de la cadena. Los trifosfatos de abacavir y lamivudina exhiben significativamente menos afinidad por las ADN polimerasas de las células huésped. En un estudio de 20 pacientes infectados con VIH que recibían 300 mg de abacavir 2 veces al día, con sólo una dosis de 300 mg tomada antes del período de muestreo de las 24 horas, la media geométrica de la vida media intracelular del trifosfato de carbovir en estado estable fue de 20.6 horas, en comparación con la media geométrica de la vida media plasmática del abacavir en este estudio de 2.6 horas. Se realizó una comparación entre las propiedades farmacocinéticas en estado estacionario de una dosis de 600 mg de abacavir, administrados 1 vez al día, y una dosis de 300 mg de abacavir, administrados 2 veces al día, en un estudio entrecruzado que se realizó en 27 pacientes infectados con el VIH. En células sanguíneas mononucleares periféricas, el grado de exposición intracelular al trifosfato de carbovir fue mayor en los pacientes que recibieron tratamiento con 600 mg de abacavir, administrados 1 vez al día, con respecto a los valores de ABC24,ss (32% mayor), Cmax 24,ss (99% mayor) y concentraciones mínimas (18% mayores), en comparación con el régimen terapéutico de 300 mg administrados 2 veces al día. En los pacientes que estaban recibiendo 300 mg de lamivudina 1 vez al día, la vida media intracelular terminal del TF de lamivudina exhibió una prolongación de 16 19 horas, en comparación con la vida media de la lamivudina plasmática de 5-7 horas. Se realizó una comparación entre las propiedades farmacocinéticas en estado estacionario de una dosis de 300 mg de lamivudina, administrados 1 vez al día durante 7 días, y una dosis de 150 mg de lamivudina, administrados 2 veces al día durante 7 días, en un estudio entrecruzado que se realizó en 60 voluntarios sanos. En células sanguíneas mononucleares periféricas, el grado de exposición intracelular al trifosfato de lamivudina fue similar en lo concerniente al ABC24,ss y Cmax 24,ss; sin embargo, las concentraciones mínimas fueron menores en comparación con el régimen terapéutico de 150 mg administrados dos veces al día. Las concentraciones intracelulares de trifosfato de lamivudina exhibieron una mayor variabilidad, en comparación con las concentraciones plasmáticas mínimas de lamivudina. Estos datos apoyan el uso de 300 mg de lamivudina y 600 mg de abacavir 1 vez al día para el tratamiento de los pacientes infectados con el VIH. Además, la eficacia y la seguridad de esta combinación administrada 1 vez al día han sido demostradas en un estudio clínico cardinal (CNA30021- ver Estudios Clínicos). Efectos farmacodinámicos: Se ha demostrado que la lamivudina tiene un alto grado de sinergismo con la zidovudina, al inhibir la replicación del VIH en cultivo de células. El abacavir exhibe sinergismo in vitro en combinación con el amprenavir, la nevirapina y la zidovudina. Se ha demostrado que es aditiva en combinación con la didanosina, zalcitabina, estavudina y lamivudina. La resistencia del VIH 1 a la lamivudina entrańa el desarrollo de un cambio del aminoácido M184V cercano al sitio de actividad de la TI viral. Esta variante surge tanto in vitro como en los pacientes infectados con el VIH 1 tratados con terapia antirretroviral que contenga lamivudina. Las mutaciones M184V exhiben susceptibilidad considerablemente disminuida a la lamivudina así como decremento de su capacidad replicativa viral in vitro. Los estudios in vitro indican que los aislados de virus resistentes a la zidovudina pueden volverse sensibles a este antirretroviral cuando adquieren simultáneamente resistencia a la lamivudina. Sin embargo, la relevancia clínica de esos hallazgos todavía no ha sido bien definida. Se han seleccionado in vitro aislados del VIH 1 resistentes al abacavir y están asociados con cambios genotípicos específicos en la región de los codones (codones M184V, K65R, L74V y Y115F) de la transcriptasa inversa (TI). La resistencia viral al abacavir se desarrolla en forma relativamente lenta in vitro e in vivo, y requiere varias mutaciones para alcanzar un aumento de ocho veces de la concentración inhibitoria 50% (CSI50%) sobre el virus de tipo silvestre, el cual podría ser un nivel clínicamente relevante. Los aislados resistentes al abacavir también pueden exhibir sensibilidad disminuida a la lamivudina, la zalcitabina, el tenofovir, la emtricitabina y/o la didanosina, pero siguen siendo sensibles a la zidovudina y la estavudina. Es improbable que llegue a presentarse resistencia cruzada entre el abacavir o la lamivudina y los antirretrovirales de otras clases; por ejemplo, inhibidores de la proteasa (IPs) o inhibidores no nucleosídicos de la transcriptasa inversa (INNTIs). Se ha demostrado decremento de la susceptibilidad al abacavir en los aislados clínicos de pacientes con replicación viral no controlada, que habían sido tratados previamente con otros inhibidores nucleosídicos y eran resistentes a los mismos. Es improbable que los aislados clínicos con tres o más mutaciones asociadas con los INTIs sean susceptibles al abacavir. La resistencia cruzada conferida por la mutación M184V de la TI está limitada a la clase de agentes antirretrovirales designados como inhibidores nucleosídicos. La zidovudina, la estavudina, el abacavir y el tenofovir mantienen sus actividades antirretrovirales contra el VIH 1 resistente a la lamivudina que sólo albergan la mutación M184V. Farmacocinética: Se ha demostrado que el comprimido de Kivexa es bioequivalente al abacavir y la lamivudina administrados por separado. Esto fue demostrado en un estudio de bioequivalencia de dosis únicas, con intercambio cruzado trilateral de grupos, de Kivexa (en ayunas) contra 2 x comprimidos de 300 mg de abacavir más 2 x comprimidos de 150 mg de lamivudina (en ayunas) contra Kivexa administrada con una comida con alto contenido de grasa, en voluntarios sanos (n=30). En el estado de ayuno no hubo diferencias significativas en el grado de absorción de cada componente, determinado por medio de la curva del área de la concentración plasmática tiempo (ABC) y la concentración máxima (Cmax). Tampoco se observaron efectos clínicamente significativos de los alimentos entre la administración de Kivexa en estado de ayuno o en estado posprandial. Estos resultados indican que Kivexa puede tomarse con o sin alimentos. A continuación se describen las propiedades farmacocinéticas de la lamivudina y el abacavir. Absorción: Después de la administración oral, el abacavir y la lamivudina son absorbidos en forma rápida y satisfactoria. La biodisponibilidad absoluta del abacavir y de la lamivudina orales en los adultos es de 83 y 80-85%, respectivamente. El tiempo medio hasta las máximas concentraciones séricas (tmax) es de aproximadamente 1.5 y 1.0 horas para el abacavir y la lamivudina, respectivamente. Después de una sola dosis oral de 600 mg de abacavir, la Cmax media es de 4.26 mcg/ml y el área bajo la curva del tiempo 0 extrapolado al infinito (ABCĄ) es de 11.95 mcg.h/ml. Después de la administración de dosis orales repetidas de 300 mg de lamivudina 1 vez al día durante siete días, los valores de las medias de la Cmax en estado estable son de 2.04 mcg/ml y los del área bajo la curva de 24 horas (media ABC24) son de 8.87 mcg.h/ml, respectivamente. Distribución: Los estudios de administración intravenosa con abacavir y lamivudina demostraron que el volumen aparente de distribución medio es de 0.8 y 1.3 l/kg, respectivamente. Los estudios de fijación a las proteínas plasmáticas in vitro indican que el abacavir sólo se fija en un grado bajo a moderado (~49%) a las proteínas plasmáticas humanas cuando se encuentra en concentraciones terapéuticas. La lamivudina exhibe una farmacocinética lineal en el intervalo posológico terapéutico así como un bajo grado de fijación a las proteínas plasmáticas (menos de 36%). Esto indica una baja probabilidad de interacciones con otros productos medicinales por desplazamiento de la fijación a las proteínas plasmáticas. Los datos demuestran que el abacavir y la lamivudina penetran el sistema nervioso central (SNC) y llegan al líquido cefalorraquídeo (LCR). Los estudios con abacavir demuestran una relación de las ABCs de las concentraciones en el LCR/plasma de entre 30 y 44%. Los valores observados de las concentraciones máximas son 9 veces mayores que la concentración inhibitoria media (CI50) del abacavir, de 0.08 mcg/ml o 0.26 micromol, cuando este antirretroviral se administra en dosis de 600 mg 2 veces al día. La relación media de las concentraciones de lamivudina en el LCR/suero 2 a 4 horas después de la administración oral fue de aproximadamente 12%. Se desconoce el grado verdadero de la penetración de la lamivudina en el SNC y su relación con cualquier eficacia clínica. Metabolismo: El abacavir es metabolizado principalmente por el hígado y menos de 2% de la dosis administrada es excretado por la ruta renal, como el compuesto intacto. Las principales rutas de metabolismo en el hombre son por la ruta de la alcohol deshidrogenasa y por glucuronidación para producir el ácido 5-carboxílico y 5-glucurónido los cuales representan alrededor de 66% de la dosis administrada. Estos metabolitos son excretados en la orina. El metabolismo de la lamivudina es una ruta de eliminación poco importante. La lamivudina es eliminada predominantemente en forma intacta por excreción renal. La probabilidad de interacciones metabólicas con la lamivudina es baja, debido a que sólo un pequeńo porcentaje (menos de 10%) experimenta metabolismo hepático. Eliminación: La vida media promedio del abacavir es de aproximadamente 1.5 horas. Después de la administración de dosis orales repetidas de 300 mg de abacavir 2 veces al día, no hay acumulación significativa del abacavir. La eliminación del abacavir tiene lugar por metabolismo hepático con excreción subsecuente de sus metabolitos principalmente en la orina. Los metabolitos y el abacavir intacto representan alrededor de 83% de la dosis eliminado en la orina. El resto es eliminado en las heces. La vida media de eliminación observada de la lamivudina es de 5 a 7 horas. El aclaramiento sistémico medio de la lamivudina es de aproximadamente 0.32 L/h/kg, predominantemente por aclaramiento renal (más de 70%) a través del sistema de transporte catiónico orgánico. Poblaciones especiales de pacientes: Pacientes con insuficiencia hepática: Se han obtenido datos farmacocinéticos del abacavir y la lamivudina en forma individual. El abacavir es metabolizado principalmente por el hígado. Los parámetros farmacocinéticos del abacavir han sido estudiados en pacientes con insuficiencia hepática leve (calificación de Child Pugh de 5 a 6). Los resultados demostraron que hubo un aumento medio de 1.89 veces en el ABC de abacavir y de 1.58 veces en la vida media del abacavir. Las ABC de los metabolitos no fueron modificadas por la enfermedad hepática. Sin embargo, las velocidades de formación y eliminación de los metabolitos disminuyeron. Es probable que se requiera reducir la dosis de abacavir en los pacientes con insuficiencia hepática leve. Por lo tanto, para tratar a estos pacientes debería usarse la preparación separada de abacavir (Ziagen). Los parámetros farmacocinéticos del abacavir no han sido estudiados en pacientes con insuficiencia hepática moderada o severa. Se espera que las concentraciones plasmáticas del abacavir sean variables y con aumentos sustanciales en estos pacientes. Por lo tanto, Kivexa está contraindicado en pacientes con insuficiencia hepática moderada y severa. Los datos obtenidos acerca de la lamivudina en pacientes con insuficiencia hepática moderada a severa demuestran que los parámetros farmacocinéticos no son afectados significativamente por la disfunción hepática. Pacientes con insuficiencia renal: Se han obtenido datos farmacocinéticos del abacavir y la lamivudina en forma individual. El abacavir es metabolizado principalmente por el hígado y alrededor de 2% del abacavir es excretado en forma intacta en la orina. La farmacocinética del abacavir en los pacientes con enfermedad renal es semejante a la de los pacientes con función renal normal. Los estudios con lamivudina demuestran que las concentraciones plasmáticas (ABC) son más altas en los pacientes con disfunción renal, debido al aclaramiento disminuido. Es necesario reducir la dosis para los pacientes con una cleareance de creatinina menor de 50 mL/minuto; por lo tanto, para tratar a estos pacientes debería usarse la preparación separada de lamivudina (Epivir). Estudios clínicos: El abacavir y la lamivudina han sido utilizados como componentes de la terapia antirretroviral de combinación en pacientes sin y con exposición previa a terapia antirretroviral. La terapia de combinación ha consistido en otros agentes antirretrovirales de la misma clase o de diferentes clases, tales como IPs y INNTIs. Se ha demostrado que el abacavir y la lamivudina de la tableta Kivexa son bioequivalentes al abacavir y la lamivudina cuando se administran por separado (ver Farmacocinética). La eficacia clínica de la terapia antirretroviral de combinación que contiene abacavir más lamivudina, administrada una o dos veces al día, ha sido confirmada en los estudios que se describen a continuación. En un estudio multicéntrico, doble ciego, controlado (CNA30024), 654 pacientes infectados con el VIH, sin exposición previa a la terapia antirretroviral, fueron aleatorizados a recibir ya fuesen 300 mg de abacavir dos veces al día o 300 mg de zidovudina dos veces al día, tanto en combinación con 150 mg de lamivudina dos veces al día como con 600 mg de efavirenz una vez al día. La duración del tratamiento doble ciego fue de por lo menos 48 semanas. En la población de Intención de Tratar (IDT), 70% de los pacientes en el grupo de abacavir, en comparación con 69% de los del grupo de zidovudina, obtuvieron una respuesta virológica en la semana 48 con concentraciones plasmáticas de ARN del VIH 1 de 50 o menos copias/ml. Los pacientes fueron estratificados de acuerdo con las concentraciones plasmáticas en la evaluación basal de ARN del VIH 1 de 100,000 copias o menos/ml o más de 100,000 copias/ml . Se demostró que el grupo de abacavir fue no inferior en comparación con el grupo de zidovudina en los subgrupos según carga viral total y basal. Este estudio confirma la no inferioridad de un régimen que contiene abacavir más lamivudina, en comparación con un régimen utilizado más extensamente de zidovudina más lamivudina. Un régimen de abacavir y lamivudina una vez al día fue investigado en un estudio multicéntrico, doble ciego, controlado (CNA30021) de 770 adultos infectados con el VIH sin exposición previa a terapia antirretroviral. Los pacientes fueron aleatorizados a recibir ya fuesen 600 mg de abacavir una vez al día o 300 mg dos veces al día, ambos en combinación con 300 mg de lamivudina una vez al día y 600 mg de efavirenz una vez al día. Los pacientes fueron estratificados en la evaluación basal de acuerdo con las concentraciones plasmáticas menores que o iguales a 100,000 ó mayores de 100,000 copias de ARN del VIH 1/mL. La duración del tratamiento doble ciego fue de por lo menos 48 semanas. Los resultados se resumen en la siguiente tabla:
Ver Tabla
Se demostró que el grupo de abacavir 1 vez al día fue no inferior cuando se comparó con el grupo de 2 veces al día en los subgrupos según carga viral total y basal. Las frecuencias de los eventos adversos reportados fueron semejantes en los dos grupos de tratamiento. Se intentó hacer un análisis genotípico de todos los sujetos con fracaso virológico (concentraciones confirmadas de ARN del VIH/mayores de 50 copias/ml). Hubo una baja frecuencia global de fracaso virológico tanto en el grupo de tratamiento de 1 vez al día como en el de 2 veces al día (10 y 8%, respectivamente). Además, por razones técnicas, la genotipificación estuvo restringida a las muestras con concentraciones plasmáticas de ARN del VIH 1/mayores de 500 copias/ml. Estos factores resultaron en un tamańo de muestra reducido. Por lo tanto, no pudieron inferirse conclusiones firmes acerca de las diferencias en las mutaciones surgidas durante el tratamiento entre los dos grupos. El residuo aminoácido de la transcriptasa inversa 184 fue consistentemente la posición más frecuente para las mutaciones asociadas con la resistencia a los INTI (M184V o M184I). La segunda mutación más frecuente fue la L74V. Las mutaciones Y115F y K65R fueron poco comunes. Datos de seguridad preclínica: No hay datos disponibles sobre los efectos de la combinación de abacavir y lamivudina en animales. Mutagenicidad y carcinogenicidad: Ni el abacavir ni la lamivudina fueron mutagénicos en las pruebas bacterianas, pero como varios análogos nucleosídicos exhiben actividad en las pruebas de células de mamífero in vitro tales como en ensayo en el linfoma de ratón. Esto concuerda con la actividad conocida de otros análogos nucleosídicos. Los estudios de carcinogenicidad con administración oral de abacavir en ratones y ratas mostraron un aumento en la frecuencia de los tumores malignos y no malignos. Los tumores malignos se presentaron en la glándula prepucial de los machos y la glándula clitoral de las hembras de ambas especies, y en el hígado, la vejiga urinaria, los ganglios linfáticos y el subcutis de las ratas hembras. La mayoría de estos tumores se presentaron con la dosis más alta de abacavir de 330 mg/kg/día en los ratones y 600 mg/kg/día en las ratas. Estos niveles posológicos fueron equivalentes a 24 y 33 veces la exposición sistémica prevista en los humanos. La excepción fue el tumor de la glándula prepucial que se presentó con una dosis de 110 mg/kg. Ésta equivale a seis veces la exposición sistémica prevista en los humanos. No hay una contraparte estructural de esta glándula en los humanos. Aunque se desconoce el potencial carcinogénico en los humanos, estos datos sugieren que el beneficio clínico potencial es más importante que el riesgo carcinogénico para los humanos. La lamivudina no ha demostrado actividad genotóxica en los estudios in vivo con dosis que produjeron concentraciones plasmáticas hasta de 30 a 40 veces más altas que las concentraciones plasmáticas clínicas. Los resultados de los estudios de carcinogenicidad a largo plazo en ratas y ratones no mostraron ningún potencial carcinogénico. Toxicidad de dosis repetidas: Después de la administración de abacavir durante dos ańos en ratones y ratas se observó degeneración miocárdica leve en el corazón. Las exposiciones sistémicas fueron equivalentes a 7-24 veces la exposición sistémica prevista en los humanos. No se ha determinado la relevancia clínica de este hallazgo. Toxicología reproductiva: En los estudios de toxicidad reproductiva en animales, se demostró que el abacavir y la lamivudina atraviesan la placenta. El abacavir sólo demostró toxicidad para los embriones y fetos en desarrollo en ratas tratadas con dosis maternalmente tóxicas de 500 mg/kg/día y mayores. Esta dosis equivale a 33 veces la exposición terapéutica humana basada en el ABC. Los hallazgos consistieron en edema fetal, variaciones y malformaciones fetales, resorciones, disminución del peso corporal fetal y aumento de los alumbramientos de productos mortinatos. La dosis con la que no hubo efectos sobre el desarrollo pre o postnatal fue de 160 mg/kg/día. Esta dosis equivale a una exposición aproximadamente 10 veces mayor que la de los humanos. No se observaron hallazgos semejantes en conejos. La lamivudina no fue teratogénica en los estudios en animales, pero hubo indicaciones de un aumento de las muertes embrionarias en una fase temprana de la gestación en conejas en las que el tratamiento produjo niveles de exposición comparables a los alcanzados en el hombre. Sin embargo, no hubo evidencia de pérdida embrionaria en las ratas en las que el tratamiento produjo niveles de exposición aproximadamente 33 veces la exposición clínica (con base en la Cmax). Los estudios de fertilidad en ratas han demostrado que ni el abacavir ni la lamivudina tuvieron efectos sobre la fertilidad de los machos o las hembras.
|
|
Posología:
La terapia debe ser instaurada por un médico experimentado en el tratamiento de la infección causada por el VIH. Kivexa no debería administrarse a adultos o adolescentes que pesen menos de 40 kg porque es un comprimido de dosis fija en el que no puede reducirse la dosis. Kivexa puede tomarse con o sin alimentos. Kivexa se presenta en comprimidos de dosis fijas y no debe prescribirse para pacientes que requieran ajuste de las dosis, tales como aquellos con clearance de creatinina de menos de 50 ml/minuto o con insuficiencia hepática leve. En los casos en que se requiera discontinuación o ajuste de la dosis, se deberían administrar preparaciones separadas de abacavir (Ziagen) o lamivudina (Epivir). En estos casos el médico debería consultar la información individual de estos productos medicinales. Poblaciones: Adultos y adolescentes: La dosis recomendada de Kivexa en adultos y adolescentes es de 1 comprimido 1 vez al día. Nińos: Kivexa no se recomienda para el tratamiento de nińos menores de 12 ańos de edad pues no puede hacerse el ajuste necesario de la dosis. Los médicos deberían consultar la información individual de los productos lamivudina y abacavir. Pacientes de edad avanzada: Los parámetros farmacocinéticos del abacavir y lamivudina no han sido estudiados en pacientes mayores de 65 ańos de edad. Cuando se traten pacientes de edad avanzada, se debe tomar en cuenta la mayor frecuencia de deterioro de la función hepática, renal y cardíaca, así como los productos medicinales o las enfermedades concomitantes. Insuficiencia renal: Aunque no es necesario ajustar la dosis de abacavir en los pacientes con insuficiencia renal, se requiere reducir la dosis de lamivudina debido al decremento de su eliminación. Por lo tanto, no se recomienda el uso de Kivexa en pacientes con un clearance de creatinina de menos de 50 ml/minuto (ver Farmacocinética). Insuficiencia hepática: Es probable que se requiera una reducción de la dosis de abacavir para los pacientes con insuficiencia hepática leve. Como no es posible reducir la dosis con Kivexa, cuando esto se considere necesario debería usarse la preparación separada de abacavir. Kivexa está contraindicado en pacientes con insuficiencia hepática moderada y severa (ver Farmacocinética).
|
|
Modo de Empleo:
Ningún requerimiento especial.
|
|
Efectos Colaterales:
Kivexa contiene abacavir y lamivudina, por lo tanto podrían esperarse los eventos adversos asociados con estos fármacos. Hipersensibilidad al abacavir (ver también Advertencias): En general, en los estudios clínicos realizados antes de la introducción del procedimiento de detección del alelo HLA-B*5701, aproximadamente 5% de los sujetos que recibieron abacavir desarrollaron una reacción de hipersensibilidad, que resultó mortal en raras ocasiones. Esta reacción se caracteriza por la aparición de síntomas que indican compromiso múltiple de órganos/sistemas corporales. Casi todos los pacientes que desarrollan reacciones de hipersensibilidad tendrán fiebre y/o erupción cutánea (generalmente maculopapular o urticarial) como parte del síndrome; sin embargo, estas reacciones se han presentado sin erupción cutánea o fiebre. Los síntomas pueden presentarse en cualquier momento mientras se está siendo tratado con abacavir, pero generalmente aparecen dentro de las seis primeras semanas de iniciación del tratamiento (tiempo mediano hasta el comienzo, 11 días). A continuación se listan los signos y los síntomas de esta reacción de hipersensibilidad. Los reportados en por lo menos 10% de los pacientes con una reacción de hipersensibilidad aparecen en negritas. Piel: Exantema (generalmente maculopapular o urticarial). Tracto gastrointestinal: Náuseas, vómito, diarrea, dolor abdominal, ulceración de la boca. Vías respiratorias: Disnea, tos, dolor de garganta, síndrome de distress respiratorio del adulto, insuficiencia respiratoria. Diversos: Fiebre, fatiga, malestar general, edema, linfadenopatía, hipotensión, conjuntivitis, anafilaxia. Neurológicos/psiquiátricos: Cefalea, parestesia. Hematológicos: Linfopenia. Hígado/páncreas: Pruebas elevadas de la función hepática, insuficiencia hepática. Musculoesqueléticos: Mialgia, en raras ocasiones miólisis, artralgia, elevación de la creatina fosfocinasa. Urología: Elevación de la creatinina, insuficiencia renal. En algunos pacientes con hipersensibilidad, inicialmente se pensó que tenían enfermedad respiratoria (neumonía, bronquitis, faringitis), una enfermedad de tipo gripal, gastroenteritis o reacciones a otras medicaciones. Este retardo en el diagnóstico de hipersensibilidad ha resultado en la continuación o la reinstauración de abacavir, lo cual ha causado una reacción de hipersensibilidad más severa o incluso la muerte. Por lo tanto, el diagnóstico de reacción de hipersensibilidad debería ser considerado cuidadosamente en los pacientes que se presentan con síntomas de estas enfermedades. Si no puede excluirse la reacción de hipersensibilidad, no debe reinstaurarse Kivexa ni ningún otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir). Los síntomas relacionados con esta reacción de hipersensibilidad empeoran con la continuación del tratamiento, y generalmente remiten con la discontinuación del abacavir. La restauración del abacavir después de una reacción de hipersensibilidad resulta en un pronto retorno de los síntomas en cuestión de horas. Esta recurrencia de la reacción de hipersensibilidad puede ser más severa que la presentación inicial y puede incluir hipotensión potencialmente fatal y la muerte. Independientemente de su estatus de HLA-B*5701, los pacientes que desarrollen esta reacción de hipersensibilidad deben discontinuar Kivexa y nunca deben ser reexpuestos a Kivexa o cualquier otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir). Ha habido reportes poco frecuentes de reacciones de hipersensibilidad después de la reintroducción de abacavir, donde la interrupción estuvo precedida por un solo síntoma clave de hipersensibilidad (exantema, fiebre, malestar general/fatiga, un síntoma gastrointestinal o un síntoma respiratorio). En muy raras ocasiones se han reportado reacciones de hipersensibilidad en pacientes que han reinstaurado el tratamiento y que no habían tenido síntomas precedentes de una reacción de hipersensibilidad. En lo que concierne a varios de los otros eventos adversos reportados, no está claro si están relacionados con la sustancia activa, con la amplia variedad de productos medicinales utilizados en el tratamiento de la enfermedad causada por el VIH o si son resultado del proceso patológico subyacente. Muchos de los eventos adversos listados se presentan comúnmente (náuseas, vómito, diarrea, fiebre, somnolencia, exantema) en pacientes con hipersensibilidad al abacavir. Por lo tanto, los pacientes con cualquiera de estos síntomas deberían ser evaluados cuidadosamente para determinar la posible presencia de esta reacción de hipersensibilidad. Si Kivexa ha sido discontinuado en pacientes debido a que experimentaron uno de estos síntomas y se decide reinstaurar el abacavir, esto sólo debe hacerse bajo supervisión médica directa (ver Consideraciones especiales después de una interrupción del tratamiento con Kivexa, en Advertencias). Datos de estudios clínicos: Frecuencia: Comunes (>1/100, <1/10); Abacavir: Hipersensibilidad al fármaco, anorexia, cefalea, náuseas, vómito, diarrea, fiebre, somnolencia, fatiga; Lamivudina: Cefalea, náuseas, vómito, dolor abdominal superior, diarrea, exantema, fatiga, malestar general, fiebre. Frecuencia: Poco comunes (>1/1.000, <1/100); Lamivudina: Neutropenia, anemia, trombocitopenia, elevaciones transitorias de las enzimas hepáticas (AST, ALT). Datos postcomercialización: Además de los eventos adversos mencionados con base en los datos de los estudios clínicos, durante el uso post-aprobatorio de abacavir y lamivudina se han identificado los siguientes eventos adversos que se presentan en la tabla que aparece a continuación. Estos eventos han sido elegidos para inclusión debido a una posible relación causal con el abacavir y/o la lamivudina: Frecuencia: Comunes (>1/100, <1/10); Abacavir: Exantema (sin síntomas sistémicos) hiperlactatemia; Lamivudina: Alopecia, artralgia, miopatías, hiperlactatemia. Frecuencia: Raros (>1/10.000, <1/1.000); Abacavir: Pancreatitis, pero la relación causal con el abacavir es incierta, acidosis láctica; Lamivudina: Rabdomiólisis, elevaciones de la amilasa sérica, pancreatitis, aunque la relación causal con la lamivudina es incierta, acidosis láctica. Frecuencia: Muy raros (<1/10.000); Abacavir: Eritema multiforme, síndrome de Stevens Johnson y necrólisis epidérmica tóxica; Lamivudina: Aplasia eritrocítica pura, parestesias, se ha reportado neuropatía periférica aunque la relación causal con el tratamiento es incierta. Se ha observado redistribución/acumulación del tejido adiposo corporal en algunos pacientes que estaban recibiendo terapia antirretroviral de combinación (ver Advertencias). La frecuencia de este evento depende de varios factores, entre los cuales figura la combinación particular de fármacos antirretrovirales.
|
|
Contraindicaciones:
Kivexa está contraindicado en pacientes con hipersensibilidad conocida al abacavir o lamivudina, o a cualquiera de los excipientes. Kivexa está contraindicado en los pacientes con insuficiencia hepática moderada y severa.
|
|
Advertencias:
En esta sección se incluyen las advertencias y precauciones especiales aplicables tanto al abacavir como a lamivudina. No hay precauciones ni advertencias adicionales relevantes a Kivexa. Hipersensibilidad al abacavir (ver también Efectos Colaterales): En general, en los estudios clínicos realizados antes de la introducción del procedimiento de detección por alelo HLA-B*5701, aproximadamente 5% de los sujetos que recibieron abacavir desarrollaron una reacción de hipersensibilidad, la cual ha resultado fatal en raras ocasiones. Factores de riesgo: Los estudios han demostrado que la presencia del alelo HLA B*5701 está asociado con un aumento significativo en el riesgo de desarrollar una reacción de hipersensibilidad al abacavir. En el estudio prospectivo CNA106030 (PREDICT-1), el uso de un método de detección del alelo HLA B*5701 previo a la terapia, y la prevención subsiguiente de la administración de abacavir a pacientes con este alelo, redujo la tasa de incidencia de reacciones de hipersensibilidad clínicamente sospechosas al abacavir, de 7.8% (66 de 847) a 3.4% (27 de 803) (p<0.0001), y la tasa de incidencia de reacciones de hipersensibilidad confirmadas mediante la prueba del parche cutáneo, de 2.7% (23 de 842) a 0.0% (0 de 802) (p<0.0001). Con base en este estudio, se estima que de 48% a 61% de los pacientes que presentan el alelo HLA B*5701 desarrollará alguna reacción de hipersensibilidad durante el ciclo terapéutico de abacavir, en comparación con 0% a 4% de los pacientes que no presentan el alelo HLA B*5701. Los médicos deberán contemplar la detección de la presencia del alelo HLA B*5701 en cualquier paciente infectado por VIH que no haya sido expuesto previamente al abacavir. Se recomienda realizar la detección del alelo HLA B*5701 antes de reiniciar el tratamiento con abacavir en pacientes con estatus de HLA-B*5701 desconocido, aunque hayan tolerado previamente el abacavir (véase "Consideraciones especiales después de la interrupción de una terapia con abacavir"). No se recomienda el uso de abacavir en pacientes que tengan diagnóstico del alelo HLA B*5701, por lo cual deberá contemplarse sólo bajo circunstancias excepcionales donde el beneficio potencial exceda al riesgo, y bajo una estrecha supervisión médica. En cualquier paciente tratado con abacavir, el diagnóstico clínico de una reacción de hipersensibilidad sospechosa deberá seguir siendo la base de una toma de decisiones clínicas. Aún en ausencia del alelo HLA B*5701, es importante suspender la administración del abacavir, y no volver a exponer al paciente a este fármaco, si no es posible descartar una reacción de hipersensibilidad por motivos clínicos, debido al potencial de que se presente una reacción severa o incluso mortal. Descripción clínica: La reacción de hipersensibilidad se caracteriza por la aparición de síntomas que indican compromiso multiorgánico. La mayoría de los pacientes tienen fiebre y/o erupción cutánea como parte del síndrome. Algunos de los otros síntomas de hipersensibilidad pueden ser fatiga, malestar general, síntomas gastrointestinales, tales como náuseas, vómito, diarrea o dolor abdominal, y signos y síntomas respiratorios tales como disnea, dolor de garganta, tos y hallazgos anormales en las radiografías torácicas (predominantemente infiltrados, los cuales pueden ser localizados). Los síntomas de esta reacción de hipersensibilidad pueden presentarse en cualquier momento durante el tratamiento con abacavir, pero generalmente se presentan dentro de las seis primeras semanas de tratamiento. Los síntomas empeoran con la continuación del tratamiento y pueden ser potencialmente mortales. Estos síntomas generalmente remiten con la discontinuación del abacavir. Tratamiento clínico: Independientemente de su estatus de HLA-B*5701, cualquier paciente que desarrolle signos o síntomas de hipersensibilidad debe ponerse en contacto inmediatamente con su médico para obtener asesoramiento. Si se diagnostica una reacción de hipersensibilidad, Kivexa debe ser discontinuado de inmediato. Kivexa, o cualquier otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir), nunca debe reinstaurarse después de una reacción de hipersensibilidad, pues podrían recurrir síntomas más severos en cuestión de horas e incluir hipotensión potencialmente mortal y muerte. Para evitar un retardo en el diagnóstico y minimizar el riesgo de una reacción de hipersensibilidad potencialmente mortal, Kivexa debe ser discontinuado permanentemente si no puede excluirse la hipersensibilidad, incluso cuando haya otros diagnósticos posibles (enfermedades respiratorias, enfermedad de tipo gripal, gastroenteritis o reacciones a otras medicaciones). Kivexa, o cualquier otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir), no debe reinstaurarse aunque se presente una recurrencia de los síntomas después de la reprovocación con una o varias medicaciones alternativas. En el paquete de Kivexa se incluye una Tarjeta de Alerta con información para el paciente acerca de esta reacción de hipersensibilidad. Consideraciones especiales después de una interrupción del tratamiento con Kivexa: Independientemente del estatus de HLA-B*5701 de un paciente, si el tratamiento con Kivexa ha sido discontinuado y se está considerando la reinstauración de la terapia, la razón de la discontinuación debe ser evaluada para asegurar que el paciente no haya tenido síntomas de una reacción de hipersensibilidad. Si no puede excluirse una reacción de hipersensibilidad, no debe reinstaurarse Kivexa ni cualquier otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir). Han habido reportes poco frecuentes de reacción de hipersensibilidad después de la reintroducción de abacavir, donde la interrupción estuvo precedida por un solo síntoma clave de hipersensibilidad (erupción cutánea, fiebre, malestar general/fatiga, síntomas gastrointestinales o un síntoma respiratorio). Si se decide reinstaurar Kivexa en estos pacientes, esto debe hacerse únicamente bajo supervisión médica directa. En muy raras ocasiones se han reportado reacciones de hipersensibilidad en pacientes que han reinstaurado el tratamiento y que no habían tenido anteriormente síntomas de una reacción de hipersensibilidad. Si se decide reinstaurar Kivexa, esto sólo debe hacerse si el paciente u otras personas pueden tener fácil acceso a una atención médica apropiada. Se recomienda realizar la detección del alelo HLA-B*5701 antes de reiniciar el tratamiento con abacavir en pacientes con estatus de HLA-B*5701 desconocido, aunque hayan tolerado previamente el abacavir. No se recomienda reiniciar el tratamiento con abacavir en aquellos pacientes con resultados positivos para la prueba de detección del alelo HLA-B*5701, y sólo se debe contemplar en circunstancias excepcionales donde el beneficio potencial exceda el riesgo, y con una estrecha supervisión médica. Información esencial para los pacientes: Los médicos que prescriban Kivexa deben asegurarse que los pacientes tengan pleno conocimiento acerca de la siguiente información sobre la reacción de hipersensibilidad: Los pacientes deben ser concientizados de la posibilidad de una reacción de hipersensibilidad al abacavir que podría resultar en una reacción potencialmente mortal o incluso la muerte y que existe un mayor riesgo de que desarrollen alguna reacción de hipersensibilidad si son HLA-B*5701-positivos. Se debe informar a los pacientes que las personas HLA-B*5701-negativas también pueden experimentar reacciones de hipersensibilidad al abacavir. Por lo tanto, cualquier paciente que desarrolle signos o síntomas consistentes con una posible reacción de hipersensibilidad al abacavir, debe contactar a su médico inmediatamente. A los pacientes que sean hipersensibles al abacavir hay que recordarles que nunca deben tomar otra vez Kivexa ni ningún otro producto medicinal que contenga abacavir (por ejemplo: Ziagen, Tricivir), independientemente de su estatus de HLA-B*5701. Para evitar la reinstauración de Kivexa, a los pacientes que hayan experimentado una reacción de hipersensibilidad debe pedírseles que devuelvan a la farmacia los comprimidos restantes de Kivexa. A los pacientes que hayan discontinuado Kivexa por cualquier razón, y particularmente por posibles reacciones adversas o enfermedad, debe recomendárseles que se pongan en contacto con su médico antes de empezar a tomarlo otra vez. A cada paciente debe recordársele que lea el Folleto Anexo al Envase del Producto incluido en el paquete de Kivexa. Debe recordárseles la importancia de sacar la Tarjeta de Alerta incluida en el paquete, y llevarla siempre consigo. Acidosis láctica/Hepatomegalia grave con esteatosis: Se han reportado acidosis láctica y hepatomegalia grave con esteatosis, incluyendo casos mortales, con el uso de análogos nucleosídicos antirretrovírales, ya sean solos o en combinación, tales como el abacavir y la lamivudina. La mayoría de estos casos se ha presentado en mujeres. Entre las características clínicas que pueden indicar el desarrollo de acidosis láctica se incluyen: debilidad generalizada, anorexia y pérdida de peso corporal repentina e inexplicable, síntomas gastrointestinales y síntomas respiratorios (disnea y taquipnea). Se debe proceder con precaución cuando se administre Kivexa a cualquier paciente y particularmente a aquellos con factores conocidos de riesgo de enfermedad hepática. El tratamiento con Kivexa debe ser suspendido en cualquier paciente que desarrolle hallazgos clínicos o de laboratorio que sugieran acidosis láctica o hepatotoxicidad (que puede incluir hepatomegalia y esteatosis, aún en ausencia de elevaciones notables en los niveles de aminotransferasa). Lipodistrofia: En algunos pacientes que recibían terapia antirretroviral en combinación se han observado, separadamente o en conjunto, redistribución/acumulación de la grasa corporal, incluyendo obesidad central, aumento del tejido adiposo dorsocervical (joroba de búfalo), emaciación periférica, adelgazamiento facial, ginecomastia, elevación de las concentraciones séricas de lípidos y sanguíneas de glucosa (ver Efectos Colaterales). Aunque todos los miembros de las clases de productos medicinales designados como inhibidores de la proteasa (IP) e inhibidores nucleosídicos de la transcriptasa inversa (INTI) han estado asociados con uno o más de estos eventos adversos específicos, vinculados con un síndrome general comúnmente conocido como lipodistrofia, los datos indican que hay diferencias en el riesgo entre los miembros individuales de las respectivas clases terapéuticas. Además, el síndrome de lipodistrofia tiene una etiología multifactorial; donde, por ejemplo, el estado de la enfermedad causada por el VIH, la edad avanzada y la duración del tratamiento antirretroviral, todos posiblemente jugando un rol sinérgico importante. Por el momento se desconocen las consecuencias a largo plazo de estos eventos. El examen clínico debe incluir evaluación en busca de signos físicos de redistribución del tejido adiposo. Se debe considerar la medición de los lípidos séricos y la glucosa sanguínea. Los trastornos lipídicos deben ser manejados en la forma clínicamente apropiada. Síndrome de Reconstitución Inmunológica: En pacientes infectados por el VIH con deficiencia inmunológica severa al momento de iniciar la terapia antirretroviral (TAR), puede surgir una reacción inflamatoria a infecciones oportunistas asintomáticas o residuales y causar condiciones clínicas serias, o agravar los síntomas. Típicamente, tales reacciones se han observado en el lapso de las primeras semanas o meses de haber iniciado la TAR. Ejemplos relevantes son la retinitis por citomegalovirus, infecciones micobacterianas generalizadas y/o focales y neumonía por Pneumocystis jiroveci (P. carinii). Cualquier síntoma inflamatorio debe ser evaluado sin retraso e iniciar el tratamiento cuando sea necesario. Se ha reportado la aparición de enfermedades autoinmunes (tales como enfermedad de Graves, polimiositis y síndrome de Guillain-Barre) en el contexto de la reconstitución inmunitaria; sin embargo, el tiempo de aparición es más variable, y puede ocurrir muchos meses después del inicio del tratamiento, y algunas veces pueden ser de presentación atípica. Pacientes coinfectados con el virus de hepatitis B: El uso de lamivudina en estudios clínicos y en productos comercializados ha demostrado que algunos pacientes con enfermedad crónica causada por el virus de hepatitis B (VHB) podrían experimentar evidencia clínica o de laboratorio de hepatitis recurrente al descontinuar la lamivudina, lo cual podría tener consecuencias más graves en los pacientes con enfermedad hepática descompensada. Si Kivexa es descontinuado en pacientes co-infectados con el virus de la hepatitis B, se deberían considerar el monitoreo periódico de las pruebas de la función hepática así como de los marcadores de la replicación del VHB. Infecciones oportunistas: Los pacientes que reciben Kivexa o cualquier otro tratamiento antirretroviral todavía pueden desarrollar infecciones oportunistas y otras complicaciones de la infección causada por el VIH. Por lo tanto, los pacientes deben permanecer bajo una estrecha observación clínica de médicos experimentados en el tratamiento de estas enfermedades asociadas con el VIH. Transmisión de la infección: Se les debe notificar a los pacientes que el tratamiento antirretroviral actual, incluso con Kivexa, no ha demostrado evitar el riesgo de transmisión del VIH a otros por contacto sexual o contaminación de la sangre. Se deben seguir tomando las precauciones apropiadas. Infarto de miocardio: En un estudio epidemiológico de carácter prospectivo y observacional, diseńado para investigar el índice de casos de infartos de miocardio en pacientes bajo terapia antirretroviral de combinación de fármacos, el uso de abacavir dentro de los seis meses previos estuvo correlacionado con un incremento en el riesgo de desarrollar infarto de miocardio. En un análisis global de estudios clínicos patrocinados por GSK, no se observó riesgo excedente alguno de desarrollar infarto de miocardio con el uso de abacavir. No existe un mecanismo biológico conocido que explique un incremento potencial. Todos los datos disponibles a partir de cohortes observacionales y estudios clínicos controlados no son concluyentes en lo que respecta a la relación entre el tratamiento con abacavir y el riesgo de desarrollar infarto de miocardio. Como medida precautoria, se deberá contemplar el riesgo subyacente de cardiopatía coronaria al prescribir terapias antirretrovirales, incluyendo abacavir, y tomar las medidas necesarias para minimizar todos los factores de riesgo modificables (por ej., hipertensión, hiperlipidemia, diabetes mellitus y tabaquismo).
|
|
Precauciones:
Embarazo y lactancia: Embarazo: No se ha establecido la seguridad del uso de Kivexa durante el embarazo humano. La lamivudina y el abacavir han estado asociados con hallazgos en los estudios de reproducción animal. Por lo tanto, la administración de Kivexa durante el embarazo sólo debería considerarse si el beneficio para la madre es más importante que el posible riesgo para el feto. Ha habido reportes de elevaciones leves y transitorias de las concentraciones séricas de lactato, las cuales podrían deberse a disfunción mitocondrial, en los neonatos y los lactantes expuestos in utero o peri partum a inhibidores nucleosídicos de la transcriptasa inversa (INTI). Se desconoce la relevancia clínica de las elevaciones transitorias del lactato sérico. En muy raras ocasiones también ha habido reportes de retardo del desarrollo, convulsiones y otra enfermedad neurológica. Sin embargo, no se ha establecido una relación causal entre estos eventos y la exposición a INTI in utero o peri partum. Estos hallazgos no afectan las recomendaciones actuales de usar terapia antirretroviral en las mujeres embarazadas para evitar la transmisión vertical del VIH. Lactancia: Los expertos en el área de la salud recomiendan que, donde sea posible, las mujeres infectadas con el virus de la inmunodeficiencia humana (VIH) no amamanten a sus hijos para evitar la transmisión del VIH. La lamivudina es excretada en la leche materna humana en concentraciones semejantes a las que se encuentran en el suero. Se espera que el abacavir también sea secretado en la leche humana, aunque esto no ha sido confirmado. Por lo tanto, se recomienda que las madres no amamanten mientras estén recibiendo tratamiento con Kivexa. Efectos sobre la capacidad para conducir vehículos y operar maquinaria: No se han hecho estudios para investigar el efecto del abacavir o la lamivudina sobre el desempeńo en la conducción de vehículos o la capacidad para operar maquinaria. Por otra parte, no puede predecirse un efecto deletéreo sobre esas actividades con base en la farmacología de estos productos medicinales. Al considerar la capacidad del paciente para conducir vehículos u operar maquinaria se deben tener presentes el estado clínico del paciente y el perfil de eventos adversos de Kivexa.
|
|
Interacciones Medicamentosas:
Como Kivexa contiene abacavir y lamivudina, todas las interacciones que puedan haberse identificado con estos agentes individualmente pueden presentarse con Kivexa. Los estudios clínicos han demostrado que no hay interacciones clínicamente significativas entre el abacavir y lamivudina. El abacavir y la lamivudina no son metabolizados significativamente por las enzimas del citocromo P450 (tales como CYP 3A4, CYP 2C9 o CYP 2D6) ni tampoco inhiben o inducen este sistema enzimático. Por lo tanto, hay poco potencial de interacciones con productos antirretrovirales tales como inhibidores de la proteasa, análogos no nucleosídicos y otros productos medicinales metabolizados por las principales enzimas del citocromo P450. La probabilidad de interacciones metabólicas con lamivudina es baja debido a la limitación de su metabolismo y la fijación a las proteínas plasmáticas, y a su aclaramiento renal casi completo. La lamivudina es eliminada predominantemente por secreción catiónica orgánica activa. Se debería considerar la posibilidad de interacciones con otros productos medicinales administrados en forma concurrente, particularmente cuando su ruta principal de eliminación sea la renal. Interacciones aplicables al abacavir: Etanol: El metabolismo del abacavir es alterado por la administración concomitante de etanol, lo cual resulta en un aumento del área bajo la curva (ABC) de abacavir de aproximadamente 41%. Tomando en cuenta el perfil de seguridad del abacavir, estos hallazgos no se consideran clínicamente significativos. El abacavir no tiene efectos sobre el metabolismo del etanol. Metadona: En un estudio farmacocinético, la coadministración de 600 mg de abacavir 2 veces al día con metadona demostró una reducción de 35% de la concentración máxima (Cmax) del abacavir y una prolongación de 1 hora en el tiempo para alcanzar la concentración máxima (Tmax), pero el ABC permaneció inalterada. Los cambios en los parámetros farmacocinéticos del abacavir no se consideran clínicamente relevantes. En este estudio, el abacavir aumentó en 22% la eliminación sistémica media de la metadona. Este cambio no se considera clínicamente relevante para la mayoría de los pacientes; sin embargo, ocasionalmente podría requerirse reajuste de la dosis de metadona. Interacciones aplicables a la lamivudina: Trimetoprim: La administración de 160/800 mg de trimetoprim/sulfametoxazol (co-trimoxazol) produce un aumento de 40% en la exposición a la lamivudina, atribuible al componente trimetoprim. Sin embargo, a menos que el paciente tenga deterioro renal, no es necesario ajustar la dosis de lamivudina (ver Posología). La lamivudina no tiene efectos sobre los parámetros farmacocinéticos del trimetoprim o el sulfametoxazol. No se ha estudiado el efecto de la coadministración de lamivudina con dosis más altas de co-trimoxazol usadas para el tratamiento de la neumonía por Pneumocystis jiriveci (P. carinii) y la toxoplasmosis. Zalcitabina: Cuando los 2 productos medicinales se usan en forma concurrente, la lamivudina podría inhibir la fosforilación intracelular de la zalcitabina. Por lo tanto, no se recomienda el uso de Kivexa en combinación con zalcitabina.
|
|
Sobredosificación:
Síntomas y signos: Aparte de los mencionados como efectos adversos, no se han identificado síntomas o signos específicos después de la sobredosificación aguda con abacavir o lamivudina. Tratamiento: En caso de sobredosis, el paciente debe ser monitoreado para determinar la posible evidencia de toxicidad, y deben aplicarse las medidas convencionales de apoyo que sean necesarias. Como la lamivudina es dializable, podría usarse hemodiálisis continua en el tratamiento de la sobredosis, aunque esto no se ha estudiado. No se sabe si el abacavir puede ser eliminado por diálisis peritoneal o hemodiálisis.
|
|
Incompatibilidades:
No aplicable.
|
|
Conservación:
Precauciones especiales de almacenamiento: No almacenar a más de 30şC. Vida útil: La fecha de caducidad está indicada en el empaque.
|
|
Observaciones:
Componentes globales del empaque: Tarjeta de alerta: Información obligatoria para el empaque: Importante: Tarjeta de alerta: Tabletas Kivexa® (sulfato de abacavir/lamivudina) lleve siempre consigo esta tarjeta. Como Kivexa contiene abacavir, algunos pacientes que toman Kivexa pueden desarrollar una reacción de hipersensibilidad (reacción alérgica seria), la cual puede ser potencialmente mortal si se continúa el tratamiento con Kivexa. Póngase en contacto inmediatamente con su médico para obtener asesoramiento acerca de si debe dejar de tomar Kivexa en caso de: 1) Experimentar alguna erupción de la piel. 2) Tener uno o más síntomas de cuando menos DOS de los siguientes grupos: Fiebre. Falta del aire (disnea), dolor de garganta o tos. Náuseas o vómito o diarrea o dolor abdominal. Fatiga intensa o sensación adolorida en todo el cuerpo o sensación de malestar general. Si usted ha discontinuado Kivexa a causa de esta reacción, nunca debe tomar otra vez Kivexa o cualquier otra medicina que contenga abacavir (Ziagen® o Trizivir®), pues en cuestión de horas podría experimentar una reducción de su presión arterial, potencialmente mortal, o incluso la muerte. División de la caja: El siguiente texto debe incluirse en una de las divisiones de la caja: La tarjeta de alerta adjunta contiene información importante de seguridad. ˇAdvertencia!: En caso de que experimente uno o más síntomas que sugieran reacción de hipersensibilidad, póngase en contacto con su médico inmediatamente.
|
|
Presentaciones:
Envase conteniendo 30 comprimidos recubiertos.
Mayor información disponible en GlaxoSmithKline Chile Farmacéutica Ltda. Avda. Andrés Bello 2687, piso 19, Edificio del Pacífico. Las Condes, Santiago, Chile. Fono: 3829000.
|
|



{kind=link}