 |
Composición:
Cada cápsula con gránulos con recubrimiento entérico contiene: Duloxetina Base (como Clorhidrato) 40 mg. Excipientes: Sacarosa, Esferas de Sacarosa, Talco, Hipromelosa 2910, Hipromelosa Acetato Succinato, Trietilcitrato, Dióxido de Titanio, Colorante FD&C Azul N° 2, Oxido de Hierro Amarillo, Oxido de Hierro Rojo, Oxido de Hierro Negro.
|
|
Acción Terapéutica:
Urológico.
|
|
Indicaciones:
Yentreve® está indicado pra el tratamiento de incontinencia urinaria de esfuerzo (IUE) moderada o severa en mujeres.
|
|
Propiedades:
Farmacodinámica: la duloxetina es un inhibidor combinado de la recaptación de serotonina y norepinefrina. La duloxetina inhibe débilmente la recaptación de dopamina sin afinidad significativa por los receptores histaminérgicos, dopaminérgicos, colinérgicos y adrenérgicos. Estudios a dosis clínicamente relevantes en seres humanos han demostrado que duloxetina disminuye la concentración de 5-hidroxitriptamina en la sangre y disminuye la excreción urinaria de norepinefrina y sus metabolitos, compatible con el bloqueo de los procesos de captación de serotonina y norepinefrina, respectivamente. Estudios preclínicos han demostrado que duloxetina bloquea potentemente la captación de serotonina y norepinefrina pero sólo inhibe ligeramente la recaptación de dopamina. La efectividad de duloxetina en el tratamiento de la incontinencia urinaria de esfuerzo (IUE) se vincula a su inhibición de la recaptación de las neuronas presinápticas de la serotonina y norepinefrina en el sistema nervioso central (médula sacroespinal) que ocasiona niveles elevados de serotonina y norepinefrina en la hendidura sináptica. En estudios realizados en animales, esto conduce a un aumento de la estimulación nerviosa del músculo del esfínter estriado de la uretra manifestada por un aumento de 8 veces en la actividad electromiográfica (EMG) del músculo, que se produce únicamente durante la fase de almacenamiento del ciclo de micción. Se cree que un mecanismo similar en las mujeres origina contracciones más fuertes de la uretra y un tono sostenido del esfínter durante el almacenamiento de la orina y explicaría la eficacia de duloxetina en el tratamiento clínico de las mujeres que sufren de IUE. El efecto de duloxetina sobre el esfínter estriado de la uretra sólo se da en la inhibición de la recaptación dual en una molécula simple y no se duplica por la administración simultánea de 2 inhibidores de la recaptación simple separados. La eficacia de la dosis de 40 mg de Yentreve® administrada 2 veces al día como tratamiento para la incontinencia urinaria de esfuerzo (IUE) se estableció en 4 estudios doble ciego, aleatorizados, controlados con placebo que enrolaron a 1913 mujeres adultas (entre 22 y 83 ańos) con incontinencia urinaria de esfuerzo (IUE) de las cuales 958 recibieron duloxetina y 955 placebo. Las principales medidas de eficacia fueron la frecuencia de los episodios de incontinencia (FEI) del paciente que completó los diarios en tiempo real y un puntaje del cuestionario de calidad de vida específico para la incontinencia (I-QOL) en 3 estudios (Estudios I-3). Frecuencia de los episodios de incontinencia: en cada uno de los 4 estudios, el grupo de pacientes tratados con duloxetina tuvo una disminución media del 50% o más en la FEI versus el 33% observado en el grupo tratado con placebo. Estas diferencias fueron observadas en cada visita después de 4 semanas (duloxetina 54%-placebo 22%), 8 semanas (duloxetina 52%-placebo 29%) y 12 semanas (duloxetina 52%-placebo 33%) de medicación. En conjunto, el 11% de los pacientes tratados con duloxetina no registró episodios de incontinencia en su última visita. En un estudio adicional limitado a pacientes con incontinencia urinaria de esfuerzo (IUE), todas las respuestas con duloxetina fueron alcanzadas dentro de las 2 semanas. La eficacia de Yentreve® no ha sido evaluada en estudios placebo-controlado de más de 3 meses. El beneficio clínico de Yentreve,® comparado con placebo, no ha sido demostrado en mujeres con incontinencia urinaria de esfuerzo (IUE) leve (definida en estudios aleatorios como aquella con FEI < 14 por semana). En estas mujeres, Yentreve® podría no proveer más beneficio que aquel ofrecido por una conducta más conservadora. Calidad de vida: los puntajes totales de calidad de vida en incontinencia (I-QOL) mejoraron significativamente en el grupo tratado con duloxetina en comparación con aquellos que recibieron placebo (9.2 versus 5.9, p < 0.001). Asimismo, usando la escala de impresión global del paciente con respecto a la mejora (PGI-I), se encontró que de manera significativa más mujeres que utilizaron duloxetina consideraron que sus síntomas de incontinencia por esfuerzo habían mejorado en comparación con las mujeres que utilizaron placebo (64.6% versus 50.1%, p < 0.001). Yentreve® y cirugía de continencia previa: existe información limitada que sugiere que los beneficios de Yentreve® no disminuyen en mujeres con incontinencia urinaria de esfuerzo (IUE) que se han sometido previamente a una cirugía de continencia. Yentreve® y entrenamiento muscular de pelvis: durante un estudio controlado, ciego, randomizado, de 12 semanas de duración, Yentreve® demostró grandes reducciones en el FEI comparado tanto con un tratamiento con placebo como con un entrenamiento muscular de pelvis. La terapia combinada de duloxetina con entrenamiento muscular de pelvis muestra grandes mejorías tanto en la medida del uso de almohadilla como en la de las condiciones específicas de calidad de vida respecto de un tratamiento solo con Yentreve® o solo de entrenamiento muscular de pelvis. Farmacocinética: Absorción: en los seres humanos, la absorción del clorhidrato de duloxetina administrado por vía oral es buena. Las concentraciones máximas (Cmax) de duloxetina en plasma ocurren 6 horas después de la dosis. La biodisponibilidad oral absoluta de duloxetina oscila entre 31.8 y 80.2% (media de 50.2%) en hombres y mujeres cuando se les administra una cápsula de 60 mg que contiene gránulos con recubrimiento entérico al 20% (peso/agua). Los alimentos no afectan la Cmax de duloxetina. Sin embargo, retardan 4 horas (de 6 a 10 horas) el tiempo para alcanzar la concentración máxima y disminuyen ligeramente la extensión de absorción (alrededor de 11%) en las mujeres cuando se les administra un dosis oral de 40 mg. La exposición de duloxetina en plasma aumenta en proporción a la dosis por encima del rango de dosis de 20 a 60 mg 2 veces al día. Las concentraciones plasmáticas en estado de equilibrio se alcanzan usualmente después de 3 días de dosificación y son aproximadamente 1.5 veces más elevadas que aquellas que se prevén a partir de la información sobre dosis únicas. Las concentraciones mínima y máxima promedio en estado de equilibrio de duloxetina luego de la administración 2 veces al día de 40 mg son 49.0 y 66.5 ng/ml, respectivamente, en las mujeres sanas. No existe una diferencia clínicamente importante en los parámetros farmacocinéticos de las dosis de la mańana y de la noche. Distribución: el volumen aparente de distribución de duloxetina administrada por vía oral oscila entre 701 y 3800 L (percentil 5to a 95to, media de 1640 L) en los hombres y mujeres sanos, lo que indica una amplia distribución en los tejidos. Duloxetina se encuentra altamente unida (> 90%) a las proteínas en el plasma humano, uniéndose principalmente a la albúmina y a la glicoproteína alfa-1 ácida. El dańo renal o hepático no afecta la unión de la proteína plasmática de duloxetina. Metabolismo: se han determinado la biotransformación y disposición de duloxetina en seres humanos luego de la administración oral de duloxetina con etiqueta 14C. Integrada en el tiempo, duloxetina comprende aproximadamente 3% del material radioetiquetado total en el plasma, lo que indica que pasa por un extenso metabolismo a varios metabolitos. Las principales vías de biotransformación para duloxetina implican la oxidación del anillo de naftil seguida por la conjugación y mayor oxidación. Tanto el CYP2D6 como el CYP1A2 catalizan la oxidación in vitro del anillo de naftil. Los 2 metabolitos principales hallados en el plasma y la orina son el conjugado de glucoronida de 4-hidroxi duloxetina y el conjugado de sulfato de 5-hidroxi, 6-metoxi duloxetina. Se ha identificado muchos metabolitos adicionales en la orina, algunos representan sólo una vía menor de eliminación. Los principales metabolitos de la circulación no son farmacológicamente activos. Excreción: la vida media de eliminación de duloxetina oscila entre 8.1 y 17.4 horas (percentil 5to a 95to, media de 12.1 horas) y la depuración aparente en plasma oscila entre 33 y 261 L/hora (percentil 5to a 95to, media de 101 L/hora) en hombres y mujeres sanos. En la orina están presentes sólo cantidades mínimas (< 1% de la dosis) de duloxetina inalterada luego de la administración de la dosis única de duloxetina con etiqueta 14C. La mayor parte de la dosis de duloxetina (72%) se recupera en la orina como metabolitos de duloxetina y aproximadamente el 19% se recupera en las heces. Poblaciones especiales: Edad: la farmacocinética de duloxetina luego de una dosis única de 40 mg se evaluó en 12 adultas mayores sanas (entre 65 y 77 ańos) y 12 mujeres sanas más jóvenes (entre 32 y 50 ańos). No hubo diferencias en la Cmax pero el área bajo la curva (ABC) de duloxetina fue 24% más elevada y la vida media 4.3 horas más prolongada en las adultas mayores. Los análisis farmacocinéticos de la población sugieren que no existe un efecto clínicamente relevante de la edad sobre la farmacocinética de duloxetina en los pacientes con IUE (rango de edad: entre 24 y 77 ańos). La farmacocinética de duloxetina no se ha estudiado en pacientes menores de 18 ańos. No es necesario realizar un ajuste de la dosis en función de la edad del paciente (véase Posología). Condición de fumador: la biodisponibilidad de duloxetina parece ser aproximadamente 34% menor en los fumadores que en los no fumadores, aunque no se recomienda efectuar modificaciones en la dosis. Raza: no se realizó un estudio farmacocinético específico para investigar los efectos de la raza. Debido a la gran variabilidad entre los pacientes, no son posibles las diferencias clínicamente relevantes en la exposición a niveles de droga entre grupos raciales. Insuficiencia renal: los pacientes con enfermedad renal en estadio terminal (ERET) que reciben diálisis crónica intermitente tuvieron valores de la Cmax y del área bajo la curva (ABC) de duloxetina aproximadamente 2 veces mayores, en comparación con las mujeres cuya función renal es normal. En contraste, la vida media de eliminación fue similar en ambos grupos. Los datos farmacocinéticos de duloxetina son limitados en pacientes con disfunción renal leve o moderada. Los análisis farmacocinéticos (FC) de la población sugieren que la disfunción renal leve no tiene un efecto significativo en la depuración aparente de duloxetina. En el caso de los pacientes con enfermedad renal en estadio terminal que reciben diálisis intermitente, el tratamiento de la IUE no es clínicamente relevante debido a que rara vez tienen una producción de orina suficiente para sufrir de IUE (véase Posología y Precauciones). Insuficiencia hepática: pacientes con cirrosis hepática tuvieron una vida media de duloxetina substancialmente más larga y la depuración (clearance) aparente en plasma de duloxetina fue aproximadamente 15% la de los individuos sanos. Una enfermedad hepática moderada (Child Pugh Clase B) afecta la farmacocinética de la duloxetina. Comparado con pacientes sanos, la depuración aparente en plasma de duloxetina fue 79% más baja, la vida media terminal aparente de duloxetina fue 2.3 veces mas larga y el área bajo la curva (ABC) fue 3.7 veces más alta en pacientes con enfermedad hepática moderada. La farmacocinética de la duloxetina y sus metabolitos no ha sido estudiada en pacientes con insuficiencia hepática leve o severa (véase Dosis y administración contraindicaciones y Precauciones).
|
|
Posología:
La dosis recomendada de Yentreve® es de 40 mg 2 veces al día independientemente de las comidas. Después de 2 a 4 semanas de tratamiento, se debe volver a evaluar a las pacientes a fin de determinar el beneficio y la tolerabilidad de la terapia. Si las pacientes experimentasen eventos adversos molestos más allá de las 4 semanas de tratamiento, la dosis podría ser reducida. La eficacia de Yentreve® no ha sido evaluada en estudios placebo-controlados de más de 3 meses de duración. El beneficio del tratamiento debe ser reevaluado en intervalos regulares. La combinación de Yentreve® con un programa de entrenamiento del músculo de la pelvis puede ser más efectiva que cualquiera de los tratamientos por separado. Se recomienda tener en consideración un entrenamiento del músculo de la pelvis en forma concomitante. Poblaciones especiales: Pacientes con insuficiencia renal: no es necesario ajustar la dosis en el caso de los pacientes con disfunción renal media o moderada (depuración de creatinina de 30 a 80 ml/min.). Cuando se trata de pacientes con ERET que reciben diálisis intermitentes, el tratamiento de la IUE no es clínicamente relevante debido a que rara vez tienen una producción de orina suficiente para sufrir de IUE. (véase Propiedades farmacocinéticas: Poblaciones especiales y Precauciones). Pacientes con insuficiencia hepática: Yentreve® no debe ser usado en mujeres con enfermedad hepática que termina en dańo hepático (véase Propiedades farmacocinéticas: Poblaciones especiales, Precauciones y Contraindicaciones). Pacientes de edad avanzada: se recomienda precaución cuando se tratan pacientes de edad avanzada. Nińos y adolescentes: la seguridad y la eficacia de duloxetina no ha sido estudiada en este tipo de pacientes. Por lo tanto, la administración de Yentreve® en nińos y adolescentes no está recomendada. Discontinuación del tratamiento: cuando se discontinúa Yentreve® después de más de 1 semana de terapia, por lo general se recomienda que la reducción de la dosis sea progresiva (de 40 mg 2 veces al día a 40 mg 1 vez al día o 20 mg 2 veces al día) durante un período de 2 semanas antes de la discontinuación a fin de reducir al mínimo los posibles síntomas que produce la discontinuación.
|
|
Efectos Colaterales:
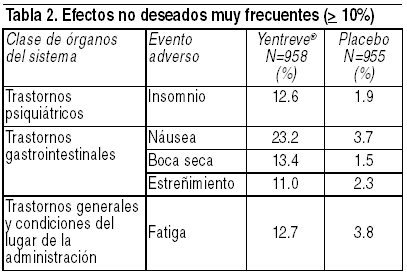
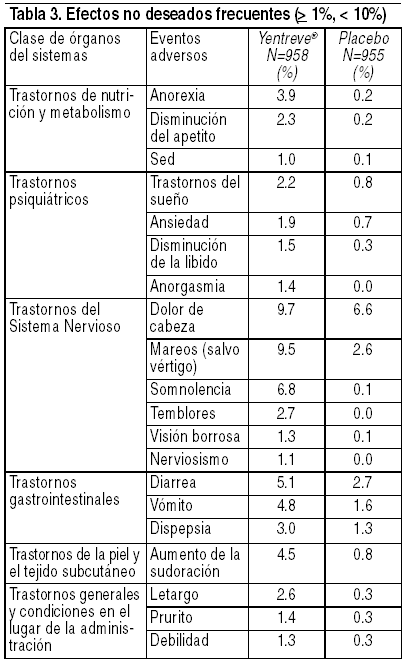
Si usted presenta alguna reacción adversa mientras esté en tratamiento con este producto, acuda a su médico. La información consignada líneas abajo se deriva de estudios clínicos fase 2-3 que incluyen a 958 pacientes tratados con duloxetina y 955 pacientes tratados con placebo en 4 pruebas controladas con placebo de 12 semanas de duración en pacientes que sufren de IUE. Esto representa 190 pacientes-ańos de exposición a 40 mg 2 veces al día. Las reacciones adversas se evaluaron mediante la recopilación de eventos adversos, resultados de exámenes físicos, signos vitales, pesos, análisis de laboratorio y electrocardiogramas. Los eventos adversos informados con más frecuencia por los pacientes tratados con Yentreve® fueron náusea, boca seca, fatiga, insomnio y estreńimiento. Los eventos adversos que se presentaron con mucho más frecuencia en pacientes que toman duloxetina en lugar de placebo y con una frecuencia de ł 2% o que fueron de potencial relevancia clínica, figuran en las tablas 2, 3 y 4. El análisis de la información mostró que el inicio de los eventos adversos informados ocurrió, por lo general, en la primera semana de la terapia. Sin embargo, la mayoría de los eventos adversos más frecuentes fueron leves a moderados y desaparecieron dentro de los 30 días siguientes a su ocurrencia (por ejemplo, la náusea). Estimado de frecuencia: muy frecuente ( ł 10%), frecuente ( ł 1% y < 10%) y poco frecuente ( ł 0.1% y < 1%).
Ver Tabla
Ver Tabla
Ver Tabla
Como se podrá apreciar en las tablas adjuntas, el 23.2% de los pacientes que recibieron duloxetina en las pruebas controladas con placebo informó acerca de náusea, el evento adverso más frecuente. Sin embargo, el 80.2% de los pacientes de duloxetina que experimentaron náusea no discontinuó el tratamiento. La mayoría de los informes de náusea producidos muy temprano luego de la iniciación de duloxetina fueron de intensidad leve o moderada (81%), no progresiva (180/181) y pasajera. Más de la mitad de los pacientes que experimentaron náusea informaron acerca de la desaparición de la misma dentro de los 7 días de haberse producido, 81% dentro de los 30 días. Asimismo, se informó acerca de casos de mareos ( ł 5%) como un evento adverso frecuente durante la discontinuación de duloxetina. Hiponatremia ha sido reportada raramente, predominantemente en pacientes de edad avanzada, cuando se administra Yentreve® y otras drogas de la misma clase farmacodinámica. Regulación de la glucosa: en estudios clínicos con duloxetina para el tratamiento del dolor neuropático diabético, el promedio de duración de la diabetes fue aproximadamente de 11 ańos, la concentración media basal de glucosa en sangre fue de 163 mg/dL y el promedio basal de la hemoglobina A1c (HbA1c) fue de 7.8%. En dichos estudios, se observaron pequeńos incrementos en los niveles de glucosa sanguínea en los pacientes tratados con duloxetina comparados con los que recibieron placebo a las 12 y 52 semanas (cuidado rutinario). El incremento fue similar en ambos períodos. Con relación al placebo o cuidado rutinario, los valores de la media de HbA1c fueron estables, no hubo aumento de peso promedio, las concentraciones medias de lípidos (colesterol, LDL, HDL, triglicéridos) fueron estables y no hubo diferencias en la incidencia de eventos adversos serios y no serios relacionados con la diabetes. Alteraciones de pruebas de laboratorio: el tratamiento con duloxetina, durante las pruebas clínicas controladas con placebo de hasta 12 semanas de duración, se asoció con pequeńos incrementos medios a partir de la línea de base al punto final en los valores de ALAT, ASAT y creatina fosfoquinasa (CPK) (2.1 U/L, 1.3 U/L y 5.8 U/L, respectivamente). Con respecto a estos analitos, se observaron valores poco frecuentes, pasajeros y anormales en las pacientes tratadas con duloxetina, en comparación con las pacientes tratadas con placebo. Cambios en los signos vitales: el tratamiento con duloxetina, durante las pruebas clínicas controladas con placebo de hasta 12 semanas, se asoció con un incremento medio a partir de la línea de base al punto final en la frecuencia del pulso en posición sentada de aproximadamente 2.4 latidos por minuto, comparado con una disminución de aproximadamente 0.5 lpm en los pacientes tratados con placebo. La incidencia de la presión sanguínea elevada sostenida (hipertensión) se examinó en los pacientes que participaron en todas las pruebas clínicas controladas con placebo. La presión sanguínea elevada sostenida se definió como una presión sanguínea sistólica en posición supina ł 140 mg Hg y un cambio a partir de la línea de base de ł 10 mm Hg, o una presión sanguínea diastólica en posición supina ł 90 mm Hg y un cambio a partir de la línea de base de ł 10 mm Hg, con cualquiera de las medidas mantenidas durante al menos 3 visitas consecutivas. No existió una diferencia significativa en la incidencia de la presión sanguínea sistólica o diastólica elevada sostenida en los pacientes que recibieron tratamiento con duloxetina comparado con los pacientes que fueron tratados con placebo. Entre los pacientes tratados con duloxetina, la incidencia de la presión sanguínea sistólica elevada sostenida fue 0.2% (2/928), mientras que la incidencia de la presión sanguínea diastólica elevada sostenida fue 0.1% (1/928). En el caso de los pacientes que recibieron placebo, la incidencia fue 0.5% (5/940) y 0.2% (2/940), respectivamente. Cambios en el peso: en las pruebas clínicas controladas con placebo, los pacientes tratados con duloxetina durante un máximo de 12 semanas experimentaron una pérdida de peso media de aproximadamente 0.3 kg, comparado con una pérdida de peso media de aproximadamente 0.1 kg en los pacientes tratados con placebo. Cambios en el electrocardiograma: se tomaron electrocardiogramas a 755 pacientes tratados con duloxetina que sufrían de IUE y a 779 pacientes tratados con placebo durante las pruebas clínicas de 12 semanas de duración. El intervalo QT corregido de la frecuencia cardíaca en las pacientes tratadas con duloxetina no fue diferente del observado en las pacientes tratadas con placebo. En resumen, esta información sugiere que no existe un potencial arritmogénico con duloxetina. Abuso y dependencia de fármacos: Sustancias de clase controlada: Duloxetina no es una sustancia controlada. Dependencia física y psicológica: en estudios realizados en animales, duloxetina no demostró potencial de abuso similar al de los estimulantes o barbitúricos (depresores). Duloxetina produjo reducciones en la actividad de roedores y monos. En estudios de farmacodependencia, duloxetina no demostró potencial para producir dependencia en monos o ratas. Si bien es cierto que duloxetina no se ha estudiado de manera sistemática en seres humanos para determinar su potencial de abuso en pacientes con incontinencia urinaria de esfuerzo, no existió un indicio de comportamiento que denote la búsqueda de la droga en las pruebas clínicas.
|
|
Contraindicaciones:
La duloxetina está contraindicada en pacientes con hipersensibilidad conocida al producto o alguno de sus excipientes. La duloxetina está contraindicada en pacientes que padecen alguna enfermedad hepática severa que genera dańo hepático (véase Propiedades farmacocinéticas: Poblaciones Especiales). Embarazo y Lactancia (véase Precauciones). No se deberá utilizar Yentreve® en combinación con inhibidores irreversibles y no selectivos de la monoaminooxidasa (IMAO) (véase Advertencias). Yentreve® no debe ser utilizado en combinación con inhibidores del CYP1A2, como fluvoxamina, ciprofloxacino y enoxacino, ya que la combinación genera concentraciones plasmáticas elevadas de duloxetina (véase Interacciones).
|
|
Advertencias:
Inhibidores de monoamino oxidasa (IMAOs).En pacientes que reciben inhibidores de la recaptación de serotonina en combinación con inhibidores de la monoamino oxidasa (IMAOs), ha habido reportes de reacciones serias, algunas veces fatales, que incluyen hipertermia, rigidez, mioclonías, inestabilidad autonómica con posibles fluctuaciones rápidas de los signos vitales, y alteraciones en la condición mental que incluyen la agitación extrema que si progresa termina en delirio y coma. Estas reacciones también han sido reportadas en pacientes que recientemente habían discontinuado el uso de inhibidores de la recaptación de serotonina y que fueron posteriormente iniciados con un IMAO. Algunos casos presentaron características semejantes al síndrome neuroléptico maligno. Los efectos del uso combinado de duloxetina e IMAOs no han sido evaluados en humanos ni en animales. Por lo tanto, ya que la duloxetina es un inhibidor tanto de la recaptación de serotonina como de la norepinefrina, se recomienda que la duloxetina no sea usada en combinación con un IMAO, o por lo menos no dentro de los 14 días de discontinuación del tratamiento con un IMAO. Basados en la vida media de la duloxetina, se debe considerar dejar pasar por lo menos 5 días luego de haber concluido el tratamiento con duloxetina antes de comenzar el tratamiento con un IMAO.
|
|
Precauciones:
Generales: Manías y convulsiones: al igual que otras drogas con mecanismos de acción similares, se deberá utilizar duloxetina con precaución en pacientes que tengan historia de manía o diagnóstico de trastorno bipolar y/o convulsiones. Midriasis: se ha informado sobre casos de midriasis en relación con el uso de duloxetina. Se deberá monitorear a los pacientes con presión intraocular elevada o aquellas que tienen riesgo de glaucoma agudo de ángulo estrecho. Uso en pacientes con enfermedades concomitantes: la experiencia clínica con duloxetina en pacientes que poseen enfermedades sistémicas concomitantes es limitada. Se debe tener precaución al administrar duloxetina a pacientes con enfermedades o condiciones que producen alteraciones en el metabolismo o respuestas hemodinámicas. Dańo hepático y renal: se produce un aumento de las concentraciones plasmáticas de duloxetina en pacientes con dańo renal severo (depuración de creatinina < 30 ml/min.) o dańo hepático severo. Se recomienda una dosis de inicio menor en este tipo de pacientes si es que es clínicamente relevante (véase Dosis y Administración, Contraindicaciones y Propiedades farmacocinéticas: Poblaciones especiales). Hemorragia: se ha informado acerca de anomalías de sangrado cutáneo tales como equimosis y púrpura con los inhibidores selectivos de la recaptación de serotonina (ISRS). Al igual que con otros agentes que inhiben la recaptación de serotonina, se aconseja tener precaución a los pacientes que toman anticoagulantes y/o medicamentos que se sabe que afectan la función plaquetaria, y a los pacientes con tendencia conocida a sangrados. Uso con antidepresivos: se debe tener precaución al utilizar Yentreve® en combinación con antidepresivos. En especial, no se recomienda la combinación con IMAOs reversibles selectivos. Sacarosa: las cápsulas duras gastrorresistentes de Yentreve® contienen sacarosa. Los pacientes con problemas de intolerancia hereditaria a la fructosa, malabsorción de glucosa-galactosa o insuficiencia de sacarosa isomaltasa no deben tomar este medicamento. Interrupción del tratamiento: algunas pacientes pueden experimentar síntomas al interrumpir el tratamiento con Yentreve,® especialmente si el tratamiento se interrumpe repentinamente. Hiponatremia: se ha comunicado raramente hiponatremia, predominante en pacientes de edad avanzada, al administrar Yentreve® y otros fármacos de la misma clase farmacodinámica. Ideas y comportamiento suicida: al igual que con otros medicamentos de acción farmacológica similar (antidepresivos), se han comunicado casos aislados de ideación y comportamiento suicida durante el tratamiento con duloxetina o poco después de la interrupción del mismo. Los médicos deben advertir a sus pacientes que les comuniquen los pensamientos o sentimientos de angustia en cualquier momento. Carcinogénesis, mutagénesis, dańo de la fertilidad: Carcinogénesis: duloxetina se administró en la dieta a ratas y ratones durante 2 ańos. En las ratas, las dosis dietarias de duloxetina de hasta aproximadamente 27 mg/kg/día en hembras (2.7 veces la dosis máxima recomendada en humanos [DMRH] sobre una base de mg/m2) o aproximadamente 36 mg/kg/día en machos (3.6 veces la DMRH sobre una base de mg/m2) no causaron un incremento en la incidencia de neoplasmas esperados o inusuales ni disminución en la latencia por algún tipo de tumor. Las ratas que recibieron concentraciones dietarias de aproximadamente 30 mg/kg/día tuvieron concentraciones de duloxetina en plasma que fueron 6 veces las concentraciones en plasma de los pacientes que recibieron la DMRH. En ratones hembra que recibieron duloxetina a aproximadamente 144 mg/kg/día (7.3 veces la DMRH sobre una base de mg/m2), se observó un incremento en la incidencia de adenomas y carcinomas hepatocelulares; sin embargo, se consideró que éstos eran secundarios a la inducción de enzimas hepáticas con hipertrofia centrolobular y vacuolación asociadas. Los ratones que recibieron concentraciones dietarias de aproximadamente 144 mg/kg/día tuvieron concentraciones de duloxetina en plasma que fueron 6 veces las concentraciones plasmáticas de los pacientes que recibieron la DMRH. Se desconoce la relevancia de esta información sobre ratones en los seres humanos. Mutagénesis: duloxetina no demostró potencial mutagénico en una serie de pruebas de genotoxicidad que incluyeron la prueba Ames de mutagénesis bacteriana, la prueba de aberración cromosómica en ovario de hámster chino (CHO), la prueba de mutagénesis en células de linfoma de ratón, la prueba de micronúcleo de ratón in vivo, la prueba de síntesis de ADN no programada de hepatocitos de rata y la prueba de intercambio de cromátidas hermanas CHO in vivo. Dańo de la fertilidad: la capacidad reproductora no resultó afectada en las ratas macho que recibieron duloxetina por vía oral en dosis de hasta 45 mg/kg/día o aproximadamente 4.6 veces la DMRH sobre una base de mg/m2. En las ratas hembra que recibieron 45 mg/kg/día de duloxetina por vía oral (4.6 veces la DMRH sobre una base de mg/m2), se demostró toxicidad reproductora debido a una disminución en el consumo de alimento materno y pérdida de peso corporal, perturbaciones en el ciclo de celo, disminución en los índices de nacimientos vivos y supervivencia de las crías y retardo en el crecimiento de las crías. El nivel de efecto no observado (NOEL) con respecto a la toxicidad de la madre, la toxicidad reproductora y la toxicidad en el desarrollo en el estudio sobre fertilidad de las hembras fue de 10 mg/kg/día (equivalente a aproximadamente la DMRH sobre una base de mg/m2). Embarazo: los estudios realizados en animales no indican efectos dańinos directos o indirectos en relación con el embarazo, desarrollo del embrión/feto, alumbramiento o desarrollo post-natal. No existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción animal no siempre pueden predecir la respuesta humana, Yentreve® deberá utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. Se deberá solicitar a las mujeres que informen a su médico si se embarazan o intentan embarazarse durante la terapia. Trabajo de parto y parto: se desconoce el efecto de duloxetina en el trabajo de parto y el parto en seres humanos. Sin embargo, ya que duloxetina y/o sus metabolitos atraviesan la placenta en ratas y debido a la posibilidad de que duloxetina y/o sus metabolitos puedan tener efectos adversos en el recién nacido, duloxetina deberá utilizarse durante el trabajo de parto y el parto sólo si el beneficio potencial justifica el riesgo potencial para el feto. Madres en período de lactancia: duloxetina y/o sus metabolitos se excretan en la leche de ratas lactantes. Se desconoce la excreción de duloxetina y/o sus metabolitos en la leche humana; sin embargo, no se recomienda la lactancia mientras se está administrando duloxetina. Uso en pediatría: no se han establecido su seguridad y su eficacia en pacientes pediátricos. Uso en geriatría: de las 958 pacientes seleccionadas en forma aleatoria en los estudios clínicos fase 2-3 de duloxetina en IUE, el 12% (111) tenía 65 ańos o más. No se observó diferencias clínicamente relevantes en la seguridad o efectividad entre estas mujeres y mujeres más jóvenes. Efectos sobre la capacidad para conducir y utilizar máquinas: no obstante en los estudios controlados sobre duloxetina no se ha demostrado que disminuya el desempeńo psicomotor, la función cognitiva o la memoria; se puede asociar con la sedación. Por consiguiente, se deberá advertir a las pacientes acerca de su capacidad para conducir un automóvil u operar maquinaria peligrosa. Conducta e ideación suicida: la posibilidad de un intento de suicidio es inherente al desorden depresivo mayor (enfermedad que podría ser tratada con duloxetina) y podría persistir hasta que ocurra una remisión significativa. Una supervisión cercana de los pacientes de alto riesgo debería acompańar a la terapia inicial. Discontinuación del tratamiento: algunos pacientes pueden experimentar síntomas de discontinuación de Yentreve® particularmente si el tratamiento es interrumpido abruptamente.
|
|
Interacciones Medicamentosas:
Drogas metabolizadas por el CYP1A2: es poco probable que duloxetina tenga un efecto clínicamente significativo sobre el metabolismo de los sustratos de CYP1A2 (por ej.: teofilina y cafeína). Los estudios de interacción farmacológica in vitro demuestran que duloxetina no induce la actividad catalítica asociada con la isoforma CYP1A2. Por consiguiente, no se prevé un incremento en el metabolismo de los sustratos CYP1A2 que resulte de la inducción, si bien no se han realizado estudios clínicos de inducción. Se ha demostrado que duloxetina es un inhibidor potencial de la isoforma CYP1A2 en estudios in vitro. Sin embargo, en un estudio clínico, la farmacocinética de teofilina, un sustrato CYP1A2, no resultó significativamente afectada por la coadministración con duloxetina (60 mg 2 veces al día). Inhibidores del CYP1A2: se aconseja tener precaución si se administra duloxetina con inhibidores del CYP1A2 (por ej.: fluvoxamina o algunos antibióticos de quinolona). Debido a que el CYP1A2 participa en el metabolismo de duloxetina, existe el potencial para un incremento en las concentraciones de duloxetina cuando se le coadministra con un inhibidor del CYP1A2. Drogas metabolizadas por el CYP2D6: se deberá tener precaución si se coadministra duloxetina con medicamentos que son metabolizados en forma predominante por el sistema CYP2D6 y que poseen un índice terapéutico muy limitado (véase Precauciones, Antidepresivos Tricíclicos) duloxetina es un inhibidor moderado de CYP2D6. Cuando se administró duloxetina a dosis de 60 mg 2 veces al día con una dosis única de desipramina, un sustrato CYP2D6, el área bajo la curva (ABC) de desipramina se triplicó. La coadministración de duloxetina (40 mg 2 veces al día) aumentó el ABC en estado de equilibrio de tolterodina (2 mg 2 veces al día) en 71% pero no afectó la farmacocinética del metabolito activo de 5-hidroxil. Inhibidores del CYP2D6: se aconseja tener precaución si se administra duloxetina con inhibidores del CYP2D6 (por ej.: los ISRS). Debido a que el CYP2D6 está involucrado en el metabolismo de duloxetina, el uso concomitante de duloxetina con inhibidores potentes de CYP2D6 puede originar concentraciones de duloxetina más elevadas. La paroxetina (20 mg cada día) disminuyó la depuración aparente en plasma de duloxetina aproximadamente 37%. Drogas que actúan en el sistema nervioso central (SNC): se aconseja tener precaución cuando se toma duloxetina en combinación con otras drogas o sustancias que actúan sobre el sistema nervioso central, especialmente aquellas de mecanismo de acción similar, incluyendo el alcohol y los sedantes (benzodiazepinas, morfinomiméticos, antipsicóticos, fenobarbital, antihistamínicos sedantes). Síndrome de serotonina: en casos poco frecuentes, se ha informado acerca del síndrome de serotonina en pacientes que utilizan ISRS en forma concomitante con medicamentos serotonérgicos. Se aconseja tener precaución si se utiliza duloxetina en forma concomitante con antidepresivos serotonérgicos como ISRS, tricíclicos como clomipramina o amitriptilina, venlafaxina o triptanol, tramadol y triptofan. Antiácidos y antagonistas de H2: la coadministración de duloxetina con antiácidos que contienen aluminio y magnesio o duloxetina con famotidina no tuvo un efecto significativo en la tasa o la extensión de la absorción de duloxetina luego de la administración de una dosis oral de 40 mg. Anticonceptivos orales y otros agentes esteroides: los resultados de varios estudios in vitro demuestran que duloxetina no induce la actividad catalítica del CYP3A, la principal vía metabólica de estos agentes. Sacarosa: las cápsulas duras gastrorresistentes de Yentreve® contienen sacarosa. Los pacientes con raros problemas hereditarios de intolerancia a la fructosa, malabsorción de la glucosa galactosa o insuficiencia a la sacarosa isomaltosa no deberán tomar esta medicina. Drogas muy unidas a la proteína plasmática: debido a que duloxetina está muy unida a la proteína plasmática (> 90%), la administración de duloxetina a un paciente que toma otra droga que está muy unida a la proteína puede causar un incremento en las concentraciones libres en cualquiera de las drogas.
|
|
Sobredosificación:
Experiencia en seres humanos: la experiencia clínica con respecto a la sobredosis de duloxetina en seres humanos es limitada. En las pruebas clínicas previas a la comercialización no se ha informado sobre casos de sobredosis aguda fatal de duloxetina. Se ha informado sobre 4 ingestiones agudas no fatales de duloxetina (300 a 1400 mg), sola o en combinación con otras drogas en pacientes que recibieron duloxetina para la depresión. Experiencia en animales: los estudios realizados en animales no ofrecen información precisa o necesariamente válida acerca del tratamiento de la sobredosis en seres humanos. Sin embargo, los experimentos realizados en animales pueden proporcionar conocimientos útiles sobre posibles estrategias de tratamiento. Se ha realizado estudios de dosis únicas con duloxetina en ratones, ratas, perros y monos. En los estudios efectuados en ratas y ratones, la dosis letal promedio (DLP) varió entre 279 y 595 mg/kg; en los estudios efectuados en perros y monos, la DLP fue > 100 mg/kg. En base a estos estudios, los principales signos de toxicidad esperados reflejan una exageración de la farmacología conocida de duloxetina. Estos incluirían efectos sobre el sistema nervioso central tales como temblores, convulsiones clónicas, ataxia, emesis y disminución del apetito. Manejo en caso de sobredosis: ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología correspondientes. En Argentina: Hospital de Pediatría Dr. Ricardo Gutiérrez: (011) 4962-6666, Hospital Dr. Alejandro Posadas: (011) 4654-6648. Optativamente otros centros de intoxicaciones. No existe un antídoto específico para duloxetina. En caso de producirse una sobredosis aguda, el tratamiento deberá constar de aquellas medidas generales empleadas en el manejo de la sobredosis de cualquier droga. Asegúrese de establecer una vía aérea, oxigenación y ventilación adecuadas. Efectúe el monitoreo del ritmo cardíaco y los signos vitales. No se recomienda inducir a emesis. Se puede indicar el lavado gástrico con un tubo orogástrico de gran calibre con protección apropiada para las vías aéreas, en caso necesario, si se realiza inmediatamente después de la ingestión. El empleo de carbón activado puede ser útil para limitar la absorción de duloxetina por parte del tracto gastrointestinal. Debido al gran volumen de distribución de esta droga, es poco probable que la diuresis forzada, la diálisis, la hemoperfusión y la transfusión de intercambio sean beneficiosas. Al momento de manejar cualquier caso de sobredosis, considere que pueden estar involucradas varias drogas. El médico deberá considerar la posibilidad de comunicarse con un centro de control de envenenamiento para obtener información adicional acerca del tratamiento de cualquier caso de sobredosis.
|
|
Conservación:
Consérvese en lugar fresco y seco a no más de 25° C. Manténgase lejos del alcance de los nińos. Manténgase las cápsulas en su envase original. No use este producto después de la fecha indicada en el envase. No repita el tratamiento sin indicación médica. No recomiende este medicamento a otra persona.
|
|
Presentaciones:
Cápsulas 40 mg: envase conteniendo 56 cápsulas con granulos.
|
|



{kind=link}
{kind=link}
{kind=link}