 |
Composición:
Cada comprimido contiene: 200 mg de Sorafenib (274 mg de Tosilato de Sorafenib).
|
|
Descripción:
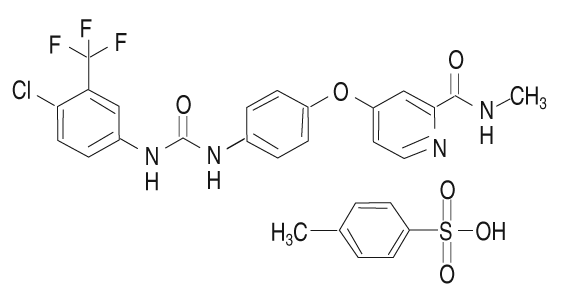
Forma farmacéutica: Comprimido redondo de color rojo, biconvexo y con recubrimiento para uso oral, con un diámetro de 10 mm y un peso de 350 mg. Grabados en bajorrelieve: En una cara: La cruz de Bayer. En la otra cara: "200". Descripción: El tosilato de sorafenib es un sólido blanco a amarillo pálido con un peso molecular de 637 g/mol. El tosilato de sorafenib es prácticamente insoluble en medios acuosos, poco soluble en etanol y soluble en PEG 400. Nombre químico: 4-metilbencenosulfonato de 4-(4-{3-[4-cloro-3-(trifluorometil)fenil]-ureido}fenoxi)-N2-metilpiridina-2-carboxiamida. La fórmula empírica es C21H16ClF3N4O3 x C7H8O3. Estructura química:
Ver Tabla
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: inhibidor de proteína cinasa. Código ATC: L01XE05.
|
|
Indicaciones:
Tratamiento de pacientes con carcinoma avanzado de células renales. Tratamiento de pacientes con carcinoma hepatocelular.
|
|
Propiedades:
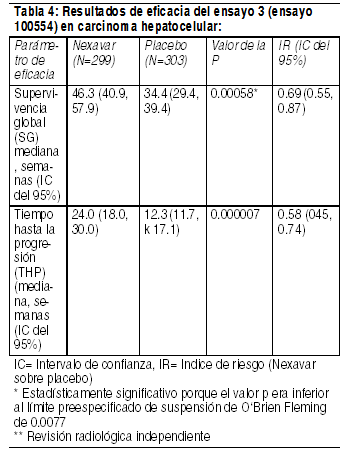
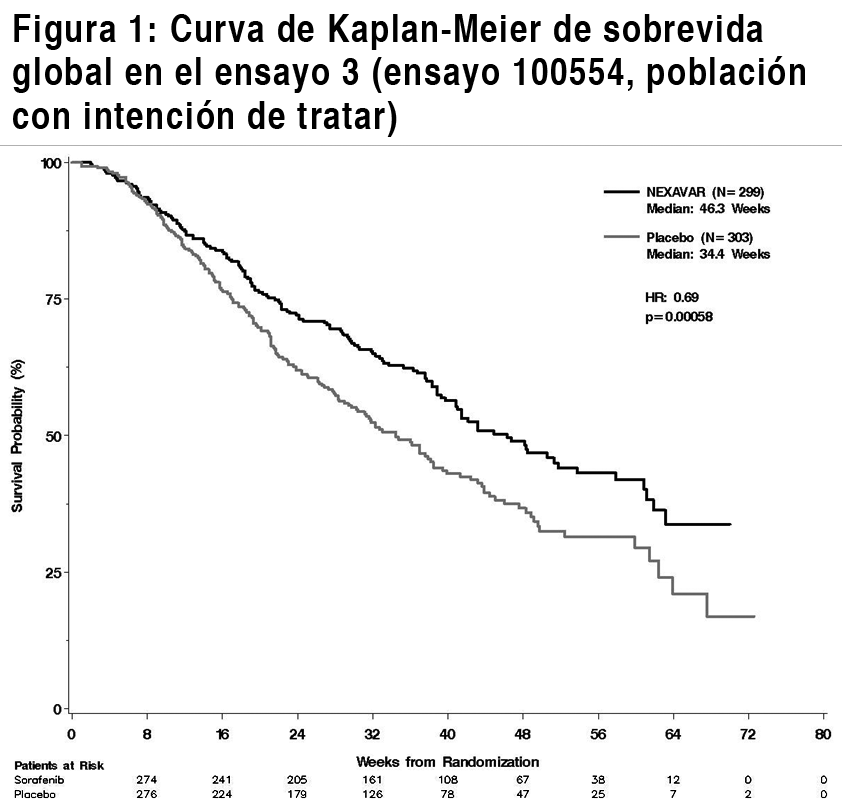
Propiedades farmacodinámicas: Mecanismo de acción: Sorafenib es un inhibidor multicinasa que reduce la proliferación celular tumoral in vitro. Se ha demostrado que sorafenib inhibe múltiples cinasas intracelulares (c-CRAF, BRAF y BRAF mutante) y de la superficie celular (KIT, FLT-3, RET, VEGFR-1, VEGFR-2, VEGFR-3 y PDGFR-ß). Varias de estas cinasas intervienen en la seńalización de las células tumorales, angiogénesis y apoptosis. Sorafenib inhibió el crecimiento tumoral del carcinoma hepatocelular y del carcinoma de células renales en humanos, así como otros xenoinjertos de tumores humanos en ratones inmunodeprimidos. En modelos de carcinoma hepatocelular y carcinoma de células renales en humanos se ha observado una disminución de la angiogénesis tumoral y un incremento de la apoptosis tumoral. Asimismo, en un modelo de carcinoma hepatocelular humano se ha observado una reducción de la seńalización de las células tumorales. Eficacia clínica: La seguridad y la eficacia clínica de Nexavar se han estudiado en pacientes con carcinoma hepatocelular (CHC) y en pacientes con Carcinoma de células renales avanzado (CCR). Carcinoma hepatocelular: El ensayo 3 (ensayo 100554) fue un ensayo de fase III, internacional, multicéntrico, aleatorio, a doble ciego, controlado con placebo en 602 pacientes con carcinoma hepatocelular. La sobrevida global (SG) fue el objetivo primario de este ensayo, mientras que el tiempo hasta la progresión (THP) fue un objetivo secundario. Las características demográficas y basales de la enfermedad eran comparables entre los grupos de Nexavar y del placebo con respecto a la edad, el sexo, la raza, el estado funcional, la etiología (incluidas las hepatitis B y C y la hepatopatía alcohólica), el estadio TNM (estadio I: <1% vs. <1%; estadio II: 10.4% vs. 8.3%; estadio III: 37.8% vs. 43.6%; estadio IV: 50.8% vs. 46.9%), la ausencia de invasión vascular macroscópica y diseminación tumoral extrahepática (30.1% vs. 30.0%) y estadio BCLC (estadio B: 18.1% vs. 16.8%; estadio C: 81.6% vs. 83.2%; estadio D: <1% vs. 0%). El estado de la función hepática según Child-Pugh era comparable entre los grupos de Nexavar y del placebo (A: 95% vs. 98%; B: 5% vs. 2%). En el ensayo sólo fue tratado un paciente con disfunción hepática C de Child-Pugh. El tratamiento previo incluyó procedimientos de resección quirúrgica (19.1% vs. 20.5%), tratamientos locorregionales (incluyendo ablación con radiofrecuencia, inyección percutánea de etanol y quimioembolización transarterial; 38.8% vs. 40.6%), radioterapia (4.3% vs. 5.0%) y tratamiento sistémico (3.0% vs. 5.0%). El ensayo se suspendió después de comprobar, en un análisis interino previsto, que la SG había traspasado la frontera preespecífica de eficacia. Este análisis de la SG mostró una ventaja estadísticamente significativa de Nexavar frente al placebo en la SG (IR: 0,69, p = 0,00058, véanse la Tabla 4 y la Figura 1). Esta ventaja fue consistente en casi todos los subgrupos analizados. En los factores preestablecidos de estratificación (categoría ECOG, presencia o ausencia de invasión vascular macroscópica y/o propagación tumoral extrahepática y región), el índice de riesgos favoreció consistentemente a Nexavar sobre el placebo. El tiempo hasta la progresión tumoral (THP, evaluado por un radiólogo independiente) era significativamente mayor en el grupo de Nexavar (IR: 0,58, p=0,000007, véase la Tabla 4).
Ver Tabla
Ver Tabla
Carcinoma de células renales: La seguridad y la eficacia de Nexavar en el tratamiento del carcinoma avanzado de células renales (CCR) se han estudiado en los 2 ensayos controlados y aleatorizados siguientes: El ensayo 1: (11213) fue un ensayo de fase III, internacional, multicéntrico, aleatorizado, a doble ciego y controlado con placebo sobre 903 pacientes. Los objetivos primarios del ensayo eran la supervivencia global y la supervivencia libre de progresión (SLP). La tasa de respuesta tumoral era un objetivo secundario. Los pacientes se aleatorizaron a Nexavar 400 mg 2 veces al día (N=45) o a placebo (N=452). Las características basales demográficas y de los pacientes estaban bien equilibradas en ambos grupos de tratamiento. Casi la mitad de los pacientes presentaba un estado funcional del ECOG de 0 y la mitad de los pacientes estaban en el grupo pronóstico bajo del MSKCC (Memorial Sloa Kettering Cancer Center). En un análisis provisional previsto de la supervivencia basado en 220 muertes, se estimó una mejoría de la supervivencia global del 39% en los pacientes tratados con sorafenib vs placebo. El índice de riesgo estimado (riesgo de muerte con sorafenib en comparación con el placebo) era 0.72 (IC del 95%: 0.55-0.95; p=0.018. El umbral de significación estadística del análisis provisional era p<0.0005). El análisis de la SLP incluyó 769 pacientes aleatorizados a Nexavar 400 mg 2 veces al día (N=384) o a placebo (N=385). La SLP se evaluó por revisión radiológica enmascarada e independiente usando los criterios RECIST. La mediana de la SLP era el doble para los pacientes aleatorizados a sorafenib (167 días), en comparación con los pacientes del placebo (84 días) (IR=0.44; IC del 95%: 0.35-055; p<0.000001). El efecto en la SLP también se exploró en diferentes subgrupos de pacientes. Los subgrupos comprendían una edad mayor de 65 ańos o menor, un estado funcional de ECOG de 0 ó 1, una categoría de riesgo pronóstico de 1 del MSKCC, si el tratamiento previo era por enfermedad metastásica progresiva o en el marco de una enfermedad anterior, y un tiempo desde el diagnóstico menor o mayor de 1.5 ańos. El efecto de sorafenib sobre la SLP fue consistente en estos subgrupos, incluyendo a los pacientes que no habían recibido tratamiento previo con IL-2 o interferón (n=137; 65 pacientes del grupo de sorafenib y 72 del grupo placebo), entre los que la mediana de la SLP era 172 días con sorafenib y de 85 días con el placebo. La mejor respuesta tumoral fue determinada por revisión radiológica del investigador conforme a los criterios RECIST. En el grupo de sorafenib, 1 paciente (0.2%) tuvo una respuesta completa, 43 pacientes (9.5%) una respuesta parcial y 333 pacientes (73.8%) tuvieron enfermedad estable. En el grupo placebo, o pacientes (0%) tuvo una respuesta completa, 8 (1.8%) tuvieron una respuesta parcial y 239 (53.9%) tuvieron enfermedad estable. Sorafenib no demostró deterioro general de los signos y síntomas específicos del cáncer renal (FKSI-10) ni de la calidad de vida relacionada con la salud, en comparación con el placebo. A las 18 y 24 semanas de tratamiento, más pacientes tratados con sorafenib comunicaron una mejoría en la puntuación total FKSI-10 (55% y 44%, respectivamente) y en la puntuación de bienestar físico (FACT-G PWB) (57% Y 47%, respectivamente) frente a placebo 8FKSI-10: 33% Y 21% y FACT-G PWB: 37% y 21%, respectivamente). El ensayo 2: Fue un ensayo de fase II, aleatorizado, de discontinuación en pacientes con tumores metastásicos, incluido CCR. La variable principal de este ensayo fue el porcentaje de pacientes aleatorizados (N=65) que permanecían sin progresión a las 24 semanas. La supervivencia libre de progresión era significativamente mayor en el grupo de sorafenib (163 días) que en el del placebo (31 días) (p=0.0001, IR=0.29). La tasa libre de progresión fue significativamente mayor en los pacientes aleatorizados a sorafenib (50%) que en los pacientes del placebo (18%) (p=0.0077). Prolongación del intervalo QT: En un ensayo de farmacología clínica se registraron las mediciones del QT/QTc en 31 pacientes en la línea basal (pre-tratamiento) y post-tratamiento. Después de un ciclo de tratamiento de 28 días, en el tiempo de la concentración máxima de sorafenib, el QTcB se prolongó en 4 ± 19 mseg y QTcF en 9 ± 18 mseg, en comparación con el tratamiento placebo en la línea basal. Ningún sujeto presentó un QTcB o QTcF > 500 mseg durante la monitorización ECG post-tratamiento (ver "Advertencias y precauciones especiales de empleo"). Propiedades farmacocinéticas: Después de la administración de los comprimidos de sorafenib, la biodisponibilidad relativa media es 38-49%, si se compara con una solución oral. La vida media de eliminación de sorafenib es de aproximadamente 25-48 horas. La administración de dosis múltiples de sorafenib durante 7 días da lugar a una acumulación de 2.5 a 7 veces en comparación con la administración de dosis únicas. Las concentraciones plasmáticas de sorafenib en estado de equilibrio se alcanzan en 7 días, con una relación de las concentraciones medias máxima a mínima menor de 2. Absorción y distribución: Después de la administración oral, sorafenib alcanza niveles plasmáticos máximos en 3 horas. Cuando se administra con una comida moderada en grasas, la biodisponibilidad es similar a la registrada en ayunas. Con una comida rica en grasas, la biodisponibilidad de sorafenib se reduce en 29% en comparación con la administración en ayunas. Los valores medios de Cmáx y AUC aumentan de manera infraproporcional a dosis superiores a 400 mg administrados oralmente 2 veces al día. La unión de sorafenib a proteínas plasmáticas humanas in vitro es del 99.5%. Metabolismo y eliminación: (En esta sección) Sorafenib se metaboliza principalmente en el hígado por metabolismo oxidativo mediado por CYP3A4, así como glucuronidación mediada por UGT1A9. Los conjugados de sorafenib pueden ser escindidos en el tracto Gl por la actividad de las glucuronidasas bacterianas, permitiendo la reabsorción del fármaco no conjugado. La coadministración de neomicina interfiere con este proceso, disminuyendo la biodisponibilidad media de sorafenib en 54%. Sorafenib representa aproximadamente 70-85% de los analitos circulantes en el plasma en estado de equilibrio. Se han identificado 8 metabolitos de sorafenib, de los que 5 se han detectado en el plasma. El principal metabolito circulante de sorafenib en plasma, el N-óxido de piridina, muestra una potencia in vitro similar a la de sorafenib y representa aproximadamente el 9-16% de los analitos circulantes en estado de equilibrio. Después de la administración oral de una dosis de 100 mg de una formulación en solución de sorafenib, el 96% de la dosis se recuperó en 14 días, con 77% de la dosis excretada en las heces y el 19% de la dosis en la orina como metabolitos glucuronizados. El sorafenib inalterado, que se supone es 51% de la dosis, se encontró en heces, pero no en orina. Ensayos de inhibición enzimática: Los ensayos con microsomas hepáticos humanos han demostrado que sorafenib es un inhibidor competitivo de CYP2C19, CYP2D6 y CYP3A4. La administración clínica concomitante de midazolam, dextrometorfano y omeprazol, que son sustratos de los citocromos CYP3A4, CYP2D6 y CYP2C19, respectivamente, después de 4 semanas de administración de sorafenib, no alteró la exposición de estos agentes. Esto indica que sorafenib no inhibe ni induce estas isoenzimas del citocromo P450. Los datos in vitro demuestran que sorafenib inhibe la glucuronidación por las vías UGT1A1 (Ki=1 µM) y UGT1A9 (Ki=2 µM). La administración clínica concomitante de sorafenib con irinotecán, cuyo metabolito activo SH-38 se sigue metabolizando por la vía UGT1A1, produjo un incremento del 67-120% del AUC de SN-38. La exposición sistémica a sustratos de UGT1A1 y UGT1A9 puede aumentar si se coadministra con sorafenib. Sorafenib inhibe CYP2B6 y CYP2C8 in vitro, con valores Ki de 6 µM y 1-2 µM, respectivamente. La administración clínica concomitante de sorafenib con paclitaxel produjo un aumento, en vez de una disminución, en la exposición de 6-OH paclitaxel, el metabolito activo de paclitaxel que se forma por CYP2C8. Estos datos sugieren que sorafenib puede no ser un inhibidor in vivo de CYP2C8. La administración concomitante de sorafenib con ciclofosfamida produjo un pequeńo aumento de la exposición a la ciclofosfamida, pero ninguna disminución de la exposición sistémica de 4-OH ciclofosfamida, el metabolito activo de la ciclofosfamida que se forma principalmente por CYP2B6. Estos datos sugieren que sorafenib puede no ser un inhibidor in vivo de CYP2B6. Los ensayos con microsomas hepáticos humanos han demostrado que sorafenib es un inhibidor competitivo de CYP2C9, con un valor Ki de 7-8 µM. Se evaluó el posible efecto de sorafenib en un sustrato de CYP2C9 en pacientes que recibían sorafenib o placebo en combinación con warfarina. Los cambios medios de los valores basales de INR-TP no eran mayores en los pacientes con sorafenib comparados con los pacientes placebo, lo que sugiere que sorafenib no puede ser un inhibidor de CYP2C9 in vivo. Efecto de los inhibidores de CYP3A4: El ketoconazol (400 mg) un potente inhibidor de CYP3A4, administrado 1 vez al día durante 7 días a voluntarios varones sanos no alteró el AUC media de una dosis única de 50 mg de sorafenib. Por tanto, no son probables interacciones farmacocinéticas clínicas entre sorafenib y los inhibidores de CYP3A4. Efecto de inductores del CYP: Las actividades CYP1A2 y CYP3A4 no se alteraron después del tratamiento de hepatocitos humanos cultivados con sorafenib, lo que indica que es improbable que sorafenib sea inductor de CYP1A2 y CYP3A4. La administración clínica concomitante continua de sorafenib y rifampicina redujo el AUC de sorafenib en un 37%, por término medio. Otros inductores de la actividad de CYP3A4 (por ej. Hypericum perforatum, también conocido como hierba de San Juan, fenitoína, carbamazepina, fenobarbital y dexametasona) también pueden aumentar el metabolismo de sorafenib y reducir, en consecuencia, las concentraciones de sorafenib. Combinación con otros agentes antineoplásicos: En ensayos clínicos se ha administrado sorafenib junto con una serie de agentes antineoplásicos en las pautas posológicas habituales incluyendo gemcitabina, oxaliplatino, paclitaxel, carboplatino, capecitabina, doxorubicina e irinotecán. Sorafenib no tuvo efectos en la farmacocinética de gemcitabina u oxaliplatino. Paclitaxel/carboplatino: La administración de paclitaxel (225 mg/m2) y carboplatino (AUC=6) con sorafenib ( Ł 400 mg 2 veces al día), administrado con una pausa posológica de 3 días en torno a la administración de paclitaxel/carboplatino, no produjo efecto significativo en la farmacocinética de paclitaxel. La coadministración de paclitaxel de (225 mg/m2, 1 vez cada 3 semanas) y carboplatino (AUC=6) con sorafenib (400 mg 2 veces al día, sin pausa en la posología de sorafenib) produjo un aumento de 47% en la exposición a sorafenib, un aumento de 29% en la exposición de paclitaxel y un aumento de 50% en la exposición de 6-OH paclitaxel. La farmacocinética de carboplatino no se afectó. Estos datos indican que no es necesario un ajuste de la dosis cuando paclitaxel y carboplatino se coadministran con sorafenib con una pausa posológica de sorafenib de 3 días. Se desconoce la significación clínica de los aumentos en la exposición de sorafenib y paclitaxel en la administración conjunta de sorafenib sin una pausa posológica. Capecitabina: La coadministración de capecitabina (750-1050 mg/m2 2 veces al día, Días 1-14 cada 21 días) y sorafenib (200 ó 400 mg 2 veces al día, administración continua ininterrumpida) no produjo cambios significativos en la exposición de sorafenib, sino un aumento de 15-50% en la exposición a capecitabina y un aumento de 0.52% en la exposición a 5-FU. Se desconoce la significación clínica de estos aumentos pequeńos a moderados en la exposición a capecitabina y 5-FU cuando se coadministraron con sorafenib. Doroxubicina/irinotecán: El tratamiento concomitante de sorafenib produjo un incremento de 21% del AUC de doxorubicina. Cuando se administró con irinotecán, cuyo metabolito activo SN-38 se sigue metabolizando por la vía UGT1A1, se produjo un incremento de 67-120% del AUC de SN-38 y un incremento del 26-42% del AUC de irinotecán. No se conoce la relevancia clínica de estos resultados (véase la sección Advertencias y precauciones especiales de empleo). Docetaxel: El docetaxel (75 ó 100 mg/m2, administrados una vez cada 21 días) coadministrado con sorafenib (200 mg 2 veces al día, o 400 mg 2 veces al día, administrados desde el día 2 al 19 de un ciclo de 21 días) con una pausa posológica de 3 días en torno a la administración de docetaxel, produjo un aumento del 36-80% en el AUC de docetaxel y del 16-32% en la Cmáx de docetaxel. Se aconseja precaución cuando se administre sorafenib con docetaxel (véase la sección Advertencias y precauciones especiales de empleo). Combinación con otros agentes: Neomicina: La coadministración de neomicina, un agente antimicrobiano no sistémico usado para erradicar flora Gl, interfiere la circulación enterohepática de sorafenib (ver más atrás), ocasionando una disminución de la exposición a sorafenib. En voluntarios sanos tratados con un régimen de 5 días de neomicina, la biodisponibilidad promedio de sorafenib disminuyó un 54%. Se desconoce la relevancia clínica de estos hallazgos. No se han estudiado los efectos de otros antibióticos, pero dependerán probablemente de su capacidad para disminuir la actividad de la glucuronidasa. Combinación con inhibidores de la bomba de protones: Omeprazol: La administración concomitante de omeprazol no afecta la farmacocinética de sorafenib. No es necesario ajustar la dosis. Farmacocinética en poblaciones especiales: Ancianos (mayores de 65 ańos) y sexo: Los análisis de los datos demográficos sugieren que no es necesario ningún ajuste posológico por edad o sexo. Pacientes pediátricos: No se dispone de datos farmacocinéticos en pediatría. Insuficiencia hepática: Sorafenib se depura principalmente a través del hígado. En pacientes con CHC e insuficiencia hepática leve (A de Child-Pugh) o moderada (B de Child-Pugh), los valores de exposición estaban en el rango observado en los pacientes sin insuficiencia hepática. La farmacocinética (PK) de sorafenib en pacientes no CHC con A de child-pugh y B de child-pugh, era similar a la PK en voluntarios sanos. La farmacocinética de sorafenib no se ha estudiado en pacientes con insuficiencia hepática grave (C de Child-Pugh) (véase la sección Advertencias y precauciones especiales de empleo y Posología). Insuficiencia renal: En un ensayo de farmacología clínica se evaluó la farmacocinética de sorafenib después de administrar una dosis única de 400 mg a sujetos con función renal normal y a sujetos con insuficiencia renal leve (ClCr 50-80 ml(min), moderada (ClCr 30 a < 50 ml/min) o grave (ClCr<30 ml/min), que no precisaban diálisis. No se observó ninguna relación entre la exposición de sorafenib y la función renal. No se precisa ningún ajuste posológico en los casos de insuficiencia renal leve, moderada o grave que no requieran diálisis (véase la sección Posología). Datos preclínicos sobre seguridad: Carcinogénesis, mutagénesis, alteración de la fertilidad: El perfil preclínico de seguridad de sorafenib se evaluó en ratones, ratas, perros y conejos. Los ensayos de toxicidad a dosis repetidas mostraron cambios de leves a moderados (degeneraciones y regeneraciones) en varios órganos. Después de la administración de dosis repetidas a perros jóvenes y en crecimiento se observaron efectos en huesos y dientes. Los cambios consistieron en un engrosamiento irregular de la placa de crecimiento femoral a una dosis diaria de sorafenib de 600 mg/m2 de superficie corporal (equivalentes a 1.2 veces la dosis clínica recomendada de 500 mg/m2 sobre la base de la superficie corporal), hipocelularidad de la médula ósea adyacente a la placa de crecimiento alterada a dosis de 200 mg/m2/día y alteraciones de la composición de la dentina a dosis de 600 mg/m2/día. Efectos similares no se indujeron en los perros adultos. Se obtuvieron efectos genotóxicos positivos con sorafenib en un ensayo de clastogenicidad (aberraciones cromosómicas) sobre células de mamífero in vitro (ovario de hámster chino) en presencia de activación metabólica. Un producto intermedio del proceso de fabricación, que también se encuentra en el producto terminado (<0.15%), fue positivo para la mutagénesis en un ensayo de células bacterianas in vitro (prueba de Ames). Sorafenib no fue genotóxico en la prueba de Ames (el material contenía un 0.34% del producto intermedio) ni tampoco en una prueba in vivo de micronúcleos de ratón. No se han realizado ensayos de carcinogenicidad con sorafenib. No se han realizado ensayos específicos con sorafenib en animales para evaluar el efecto sobre la fertilidad. Sin embargo, se puede esperar un efecto adverso sobre la fertilidad masculina y femenina porque los ensayos con dosis repetidas en animales han mostrado alteraciones en los órganos reproductores masculinos y femeninos. Las alteraciones típicas consistieron en signos de degeneración y retraso en testículos, epidídimos, próstata y vesículas seminales de las ratas; con efectos claros a una dosis diaria de sorafenib de 150 mg/m2 de superficie corporal (equivalentes a aproximadamente 0.3 veces la dosis clínica recomendada de 500 mg/m2 sobre la base de la superficie corporal). Las ratas hembra mostraron necrosis central de los cuerpos lúteos y detención del desarrollo folicular en los ovarios con un efecto menor observado a 30 mg/m2/día. Los perros mostraron degeneración tubular en los testículos a dosis de 600 mg/m2/día y oligospermia a 1200 mg/m2/día. Sorafenib ha demostrado ser embriotóxico y teratógeno cuando se administró a ratas y conejos. Los efectos observados incluyeron disminuciones de los pesos corporales maternos y fetales, un número aumentado de reabsorciones fetales y un número aumentado de malformaciones externas y viscerales. Resultados adversos fetales se observaron a una dosis oral de 6 mg/m2/día en ratas y de 36 mg/m2/día en conejos (véanse las secciones Advertencias y precauciones especiales de empleo y Embarazo y lactancia).
|
|
Posología:
Dosis recomendada: La dosis diaria recomendada de sorafenib es 400 mg (2 comprimidos de 200 mg) tomados 2 veces al día, bien sin alimentos o con una comida con un contenido bajo o moderado en grasa. Método de administración: Para vía oral. Deglutir con un vaso de agua. Duración del tratamiento: El tratamiento debe continuarse hasta que ya no suponga ningún beneficio clínico para el paciente o hasta que se manifieste una toxicidad inaceptable. Valoración de la dosis, ajuste de la dosis, recomendaciones especiales de monitorización: El tratamiento de las reacciones adversas farmacológicas sospechadas puede exigir la interrupción temporal y/o la reducción de la dosis de la terapia de sorafenib. Cuando sea necesario reducir la dosis, la dosis de sorafenib debe reducirse a 2 comprimidos de 200 mg 1 vez al día (ver la sección Advertencias y precauciones especiales). Las modificaciones posológicas propuestas en caso de toxicidad cutánea se resumen en la Tabla 0.
Ver Tabla
Poblaciones especiales: Pacientes pediátricos: No se han establecido la seguridad y la eficacia de sorafenib en pacientes pediátricos. Ancianos (mayores de 65 ańos), sexo y peso corporal: No se exige ningún ajuste posológico en base a la edad (mayores de 65 ańos), el sexo o el peso corporal del paciente. Insuficiencia hepática: No es necesario efectuar ningún ajuste posológico en pacientes con insuficiencia hepática A o B de Child-Pugh. Sorafenib no se ha estudiado en pacientes con insuficiencia hepática C de Child-Pugh (véase la sección Propiedades farmacocinéticas - Poblaciones especiales - Insuficiencia hepática). Insuficiencia renal: No necesitan ningún ajuste posológico los pacientes con insuficiencia renal leve, moderada o grave, que no precisan diálisis. Sorafenib no se ha estudiado en pacientes sometidos a diálisis (véase la sección Propiedades farmacocinéticas - Poblaciones especiales - Insuficiencia renal). Se aconseja monitorizar el balance hídrico y los electrolitos en los pacientes de riesgo de disfunción renal.
|
|
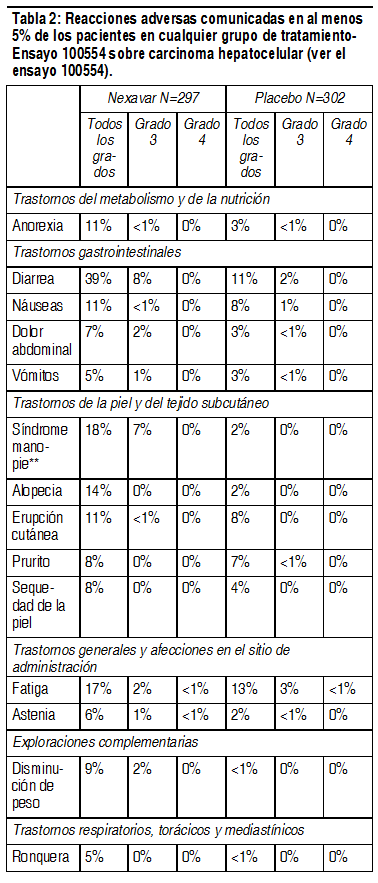
Efectos Colaterales:
Las reacciones adversas serias más importantes fueron infarto del miocardio/isquemia, perforación gastrointestinal, hepatitis inducida por el fármaco, hemorragia, e hipertensión/crisis hipertensiva. Las reacciones adversas más frecuentes fueron diarrea, erupción, alopecia y síndrome cutáneo mano-pie (corresponde al síndrome de eritrodisestesia palmoplantar de MedDRA). Las reacciones adversas reportadas en múltiples ensayos clínicos o a través del uso posterior a la comercialización se enumeran a continuación en las Tablas 1 y 2, por clase de órgano o sistema (MedDRA) y frecuencia. Las frecuencias se definen como: muy frecuentes ( ł 1/10), frecuentes ( ł 1/100 a <1/10), poco frecuentes ( ł 1/1.000 a <1/100), raras ( ł 1/10.000 a <1/1.000), desconocidas (no se pueden calcular a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Ver Tabla
Ver Tabla
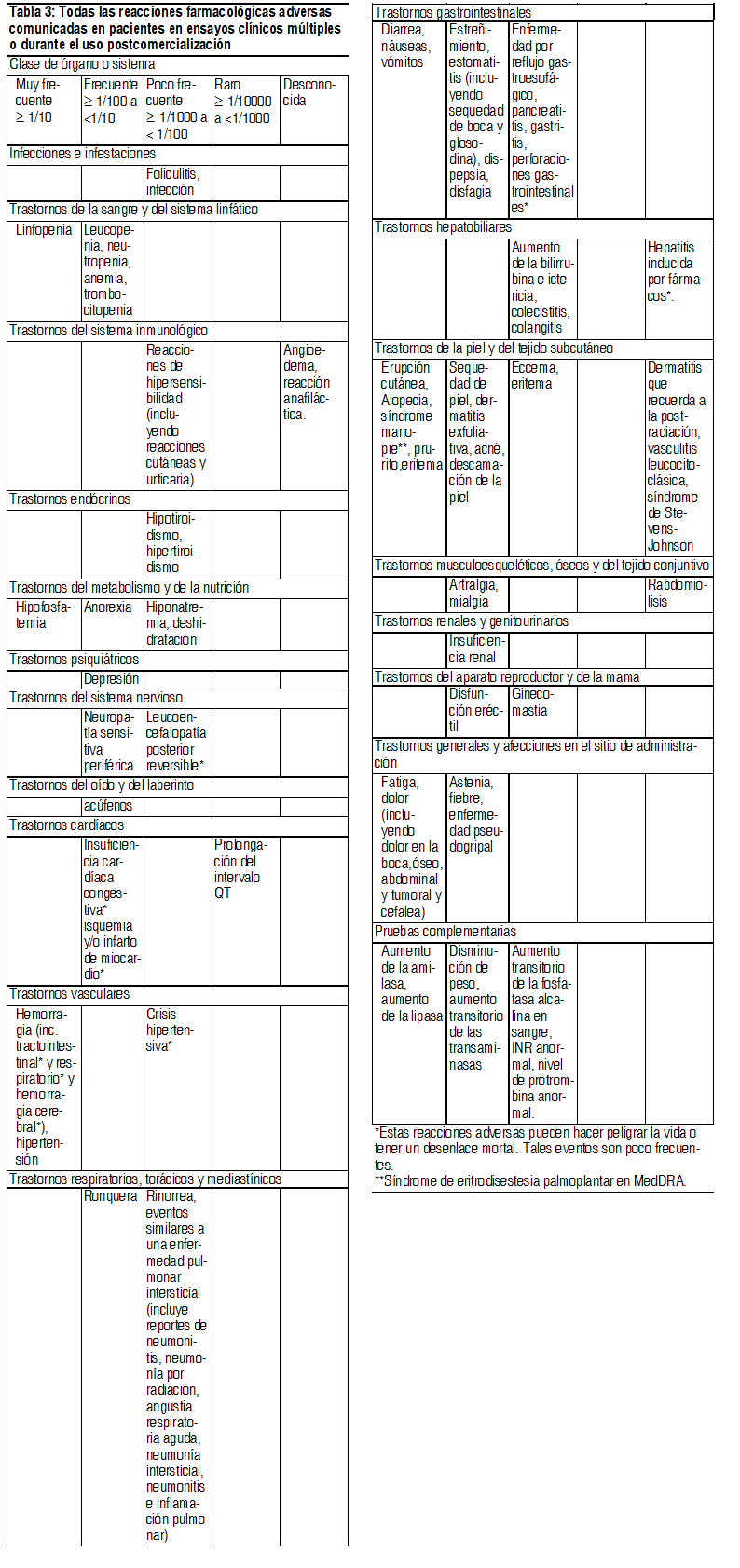
Las reacciones adversas que ocurrieron durante ensayos clínicos o se han identificado durante la postcomercialización se exponen a continuación en la Tabla 3 por clase de órgano o sistema (en MedDRA) y frecuencia. Las frecuencias se definen como: muy frecuentes ( ł 1/10), frecuentes ( ł 1/100, <1/10), poco frecuentes ( ł 1/1,000, <1/100), raras ( ł 1/10,000, <1/1,000), frecuencia desconocida (no puede estimarse de los datos disponibles). Los efectos adversos se presentan en orden decreciente de gravedad dentro de cada grupo de frecuencia.
Ver Tabla
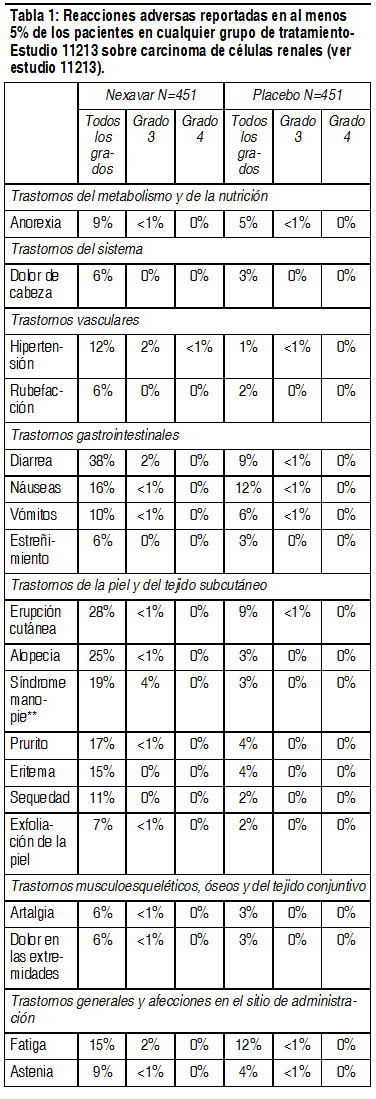
Información adicional sobre reacciones adversas farmacológicas seleccionadas: Insuficiencia cardíaca congestiva: En ensayos clínicos patrocinados por la compańía, la insuficiencia cardíaca congestiva se reportó como evento adverso en 1.9% de los pacientes tratados con sorafenib (N=2276). En el ensayo 11213 (CCR) los eventos adversos consistentes con insuficiencia cardíaca congestiva fueron reportados en 1.7% de los tratados con sorafenib y 0.7% de los tratados con placebo. En el ensayo 100554 (CHC), fueron reportados con estos eventos en 0.99% de los tratados con sorafenib y 1.1% de los tratados con placebo. Dos ensayos clínicos controlados con placebo y aleatorizados que comparaban la seguridad y eficacia de sorafenib en combinación con quimioterapias dobles basadas en platino (carboplatino/paclitaxel y separadamente gemcitabina/cisplatino) frente a las respectivas quimioterapias dobles basadas en platino, solas como tratamiento de primera línea en pacientes con carcinoma pulmonar de células no pequeńas (NSCLC) en estadio avanzado no cumplió su objetivo primario de mejorar la supervivencia global. Los eventos de seguridad fueron generalmente consistentes con los reportados previamente. Sin embargo, en ambos ensayos se observó mayor mortalidad en el subgrupo de pacientes con carcinoma pulmonar de células escamosas tratado con sorafenib y quimioterapias dobles basadas en platino frente a los tratados con quimioterapias dobles basadas en platino, solas (carboplatino/paclitaxel: IR 1.81, CI del 95% 1.19-2.74; gemcitabina/cisplatino: IR 1.22, CI del 95% 0.82 - 1.80). No se identificó ninguna causa definitiva de los hallazgos. La seguridad también se evaluó en un conjunto de ensayos de fase 2, constituido por 638 pacientes tratados con sorafenib, de ellos 202 con CCR, 137 con carcinoma hepatocelular y 299 con otros tipos de cáncer. Los eventos adversos más frecuentes relacionados con el fármaco, comunicados en los pacientes tratados con sorafenib en este conjunto, eran erupción (38%), diarrea (37%), reacción cutánea de manos y pies (35%) y fatiga (33%). Las tasas respectivas de eventos adversos relacionados con el medicamento de grado 3 y 4 de los CTC (v 2.0) en los pacientes tratados con sorafenib eran 37% y 3%, respectivamente. Anomalías en las pruebas de laboratorio de los pacientes con CCR (ensayo 11213): Con mucha frecuencia se han descrito aumento de los niveles de lipasa y amilasa. En el ensayo 11213 se produjeron aumentos de la lipasa, de grado 3 ó 4 de los CTCAE en 12% de los pacientes del grupo de sorafenib, en comparación con 7% de los del grupo placebo. Se refirieron aumentos de la amilasa de grado 3 ó 4 de los CTCAE en 1% de los pacientes del grupo de sorafenib, en comparación con 3% de los del grupo placebo. Se comunicó pancreatitis clínica en 2 de los 451 pacientes tratados con sorafenib (grado 4 de los CTCAE) así como en 1 de los 451 pacientes (grado 2 de los CTCAE) del grupo placebo del ensayo 1. La hipofosfatemia fue un hallazgo frecuente de laboratorio que se observó en 45% de los pacientes tratados con sorafenib, en comparación con 11% de los tratados con placebo. El 13% de los pacientes tratados con sorafenib y 3% de los pacientes del grupo placebo presentó hipofosfatemia (1-2 mg/dl) de grado 3 de los CTCAE). No hubo casos de hipofosfatemia de grado 4 de los CTCAE (< 1 mg/dl) en ninguno de los pacientes de sorafenib o del placebo. Se desconoce la etiología de la hipofosfatemia asociada a sorafenib. Se registró linfopenia de grado 3 ó 4 de los CTCAE en 13% de los pacientes tratados con sorafenib y en 7% de los tratados con placebo; neutropenia en 5% de los pacientes tratados con sorafenib y en 2% de los tratados con placebo; anemia en 2% de los pacientes tratados con sorafenib y en 4% de los tratados con placebo y trombocitopenia en 1% de los pacientes tratados con sorafenib y 0% en los pacientes tratados con placebo. Anomalías en las pruebas de laboratorio de los pacientes con CHC (ensayo 100554): Se observó una elevación de la lipasa en 40% de los pacientes tratados con Nexavar, en comparación con 37% de los pacientes del grupo placebo. Aumentos de la lipasa de grados 3 ó 4 de los CTCAE ocurrieron en 9% de los pacientes de cada grupo. Se observó amilasa elevada en 34% de los pacientes tratados con Nexavar, frente al 29% de los pacientes del placebo. Elevaciones de la amilasa de grados 3 ó 4 de los CTCAE se comunicaron en 2% de los pacientes de cada grupo. Muchas de las elevaciones de la lipasa y la amilasa eran transitorias y, en la mayoría de los casos, no se interrumpió el tratamiento con Nexavar. Se comunicó pancreatitis clínica en 1 de los 297 pacientes tratados con Nexavar (grado 2 de los CTCAE). Hipofosfatemia fue un hallazgo frecuente de laboratorio, observada en 35% de los pacientes tratados con Nexavar, frente al 11% de los tratados con placebo; hipofosfatemia de grado 3 de los CTCAE (1-2 mg/dl) ocurrió en 11% de los pacientes tratados con Nexavar y en 2% de los del grupo placebo; se comunicó 1 caso de hipofosfatemia de grado 4 de los CTCAE (< 1 mg/dl) en el grupo del placebo. No se conoce la etiología de la hipofosfatemia asociada a Nexavar. Las elevaciones en las pruebas de la función hepática eran comparables entre los 2 grupos del ensayo. Se observaron elevaciones de AST en 94% de los pacientes tratados con Nexavar y en 91% de los tratados con placebo; se comunicaron elevaciones de AST de grado 3 ó 4 de los CTCAE en 16% de los pacientes tratados con Nexavar frente al 17% de los tratados con placebo. Se observaron elevaciones de ALT en 69% de los pacientes tratados con Nexavar y 68% de los del placebo; elevaciones de ALT de grado 3 ó 4 de los CTCAE se comunicaron en 3% de los pacientes tratados con Nexavar y en 8% de los pacientes tratados con placebo. Se observó elevación de la bilirrubina en 47% de los pacientes tratados con Nexavar y 45% de los del placebo; elevaciones de bilirrubina de grado 3 ó 4 de los CTCAE se notificaron en 10% de los pacientes tratados con Nexavar y en 11% de los pacientes tratados con placebo. Se observó hipoalbuminemia en 59% de los pacientes tratados con Nexavar y en 47% de los pacientes del placebo; no se observó en ningún grupo hipoalbuminemia de grado 3 ó 4 de los CTCAE. Se observaron elevaciones de la fosfatasa alcalina en 82.2% de los pacientes tratados con Nexavar y en 82.5% de los tratados con placebo; elevaciones de la fosfatasa alcalina de grado 3 de los CTCAE se comunicaron en 6.2% de los pacientes tratados con Nexavar y en 8.2% de los tratados con placebo; no se observó ninguna elevación de la fosfatasa alcalina de grado 4 de los CTCAE en ningún grupo. Se observaron elevaciones de INR en 42% de los pacientes tratados con Nexavar y en 34% de los tratados con el placebo; se comunicaron elevaciones de INR de grado 3 de los CTCAE en 4% de los pacientes tratados con Nexavar y en 2% de los tratados con placebo y no hubo ningún aumento del INR de grado 4 de los CTCAE en ningún grupo. Se observó linfopenia en 47% de los pacientes tratados con Nexavar y en 42% de los tratados con placebo; se comunicó linfopenia de grado 3 ó 4 de los CTCAE en 6% de los pacientes de cada grupo. Se observó neutropenia en 11% de los pacientes tratados con Nexavar y en 14% de los pacientes placebo; se comunicó neutropenia de grado 3 ó 4 de los CTCAE en 1% de los pacientes de cada grupo. Se observó anemia en 59% de los pacientes tratados con Nexavar y en 64% de los tratados con placebo; se comunicó anemia de grado 3 ó 4 de los CTCAE en 3% de los pacientes de cada grupo. Se observó trombocitopenia en 46% de los pacientes tratados con Nexavar y en 41% de los pacientes del placebo; se comunicó trombocitopenia de grado 3 ó 4 de los CTCAE en 4% de los pacientes tratados con Nexavar y en menos de 1% de los pacientes placebo.
|
|
Contraindicaciones:
Sorafenib está contraindicado en los pacientes con hipersensibilidad grave conocida a sorafenib o a cualquiera de los excipientes.
|
|
Advertencias:
Advertencias y precauciones especiales de empleo: Embarazo: Durante el tratamiento, las mujeres deben evitar el embarazo. Las mujeres en edad fértil deben conocer los posibles riesgos para el feto, que incluyen malformaciones graves (teratogenia), retraso del crecimiento y muerte fetal (embriotoxicidad). Sorafenib no debe utilizarse durante el embarazo. El médico sólo puede considerar su uso si los posibles beneficios justifican los riesgos potenciales para el feto. En base al mecanismo propuesto de inhibición multicinasa y a los múltiples eventos adversos observados en animales expuestos a niveles significativamente inferiores a la dosis clínica, se ha de suponer que sorafenib causa dańo fetal cuando se administra a una mujer embarazada. Durante el tratamiento con sorafenib se interrumpirá la lactancia materna (véase la sección Embarazo y Lactancia y la sección Datos preclínicos sobre seguridad). Toxicidad dermatológica: Las reacciones farmacológicas adversas más frecuentes de sorafenib son reacción cutánea en manos y pies (eritrodisestesia palmoplantar) y erupción. La reacción cutánea en manos y pies y la erupción son normalmente de grado 1 y 2 de CCT (National Cancer Institute Frequent Toxicity Criteria - Criterios Comunes de Toxicidad del Instituto Nacional del Cáncer) y, en general, aparecen durante las primeras 6 semanas de tratamiento con sorafenib. El tratamiento de la toxicidad dermatológica puede incluir tratamiento tópico para el alivio sintomático, la interrupción temporal del tratamiento y/o la modificación de la dosis de sorafenib o, en casos graves o persistentes, la interrupción permanente de sorafenib (véase la sección Efectos colaterales). Hipertensión: Se observó incremento en la incidencia de hipertensión en los pacientes tratados con sorafenib. En general, la hipertensión fue de leve a moderada, tuvo lugar al principio del tratamiento y se controló con el tratamiento antihipertensivo habitual. La presión arterial debería controlarse regularmente y tratarse, en caso necesario, según la práctica médica acostumbrada. En caso de hipertensión grave o persistente, o de crisis hipertensiva a pesar de un tratamiento antihipertensivo adecuado, debiera considerarse la interrupción permanente de sorafenib (véase la sección Efectos colaterales). Hemorragia: El riesgo de hemorragia puede aumentar después de la administración de sorafenib. La incidencia de eventos hemorrágicos graves es poco frecuente. Si un evento hemorrágico precisara de intervención médica, se recomienda considerar la interrupción permanente de sorafenib (véase la sección Efectos colaterales). Warfarina: En algunos pacientes se han comunicado eventos hemorrágicos poco frecuentes o aumentos del índice internacional normalizado (INR) durante el tratamiento simultáneo de warfarina y sorafenib. Si el paciente toma warfarina concomitantemente, se controlarán regularmente los cambios en el tiempo de protrombina, INR y los episodios hemorrágicos clínicos (véase la sección Efectos colaterales). Complicaciones en la cicatrización de heridas: No se han realizado ensayos formales sobre el efecto de sorafenib en la cicatrización de heridas. En pacientes sometidos a cirugía mayor, se recomienda la interrupción transitoria del tratamiento con sorafenib como medida de precaución. Es limitada la experiencia clínica relativa al intervalo necesario para reiniciar el tratamiento tras cirugía mayor. Por tanto, la decisión de reiniciar el tratamiento con sorafenib después de cirugía mayor se basará en el juicio clínico sobre la cicatrización adecuada de la herida. Isquemia cardíaca y/o infarto: En el ensayo 11213, la incidencia de eventos de isquemia/infarto cardíaco emergentes durante el tratamiento era mayor en el grupo de sorafenib (4.9 %) que en el grupo placebo (0.4%). En el ensayo 100554, la incidencia de eventos de isquemia/infarto cardiaco emergentes durante el tratamiento era 2.7% en los pacientes tratados con sorafenib y 1.3% en el grupo placebo. Los pacientes con enfermedad arterial coronaria inestable o infarto reciente de miocardio fueron excluidos de estos ensayos. Se sopesará la interrupción temporal o permanente de sorafenib si el paciente presenta isquemia cardíaca o infarto de miocardio (véanse las secciones Efectos colaterales, Propiedades farmacocinéticas y Eficacia clínica). Prolongación del intervalo QT: Se ha demostrado que Nexavar prolonga el intervalo QT/QTc (ver Propiedades farmacológicas: - Propiedades farmacodinámicas), lo que puede ocasionar un riesgo aumentado de arritmias ventriculares. Usar sorafenib con precaución en pacientes que tienen, o pueden presentar, prolongación del QTc, como pacientes con síndrome del QT largo congénito, pacientes tratados con dosis acumulativas altas de antraciclina, pacientes que toman determinados antiarrítmicos u otros medicamentos que ocasionen prolongación del QT y los que tienen alteraciones electrolíticas como hipopotasemia, hipocalcemia o hipomagnesemia. Cuando se use Nexavar en estos pacientes se debe considerar la monitorización periódica con electrocardiogramas durante el tratamiento y electrólitos (magnesio, potasio, calcio). Perforación gastrointestinal: La perforación gastrointestinal es un evento poco frecuente y ha sido notificada en menos del 1% de los pacientes tratados con sorafenib. En algunos casos no estuvo asociada con tumor intraabdominal aparente. Deberá interrumpirse el tratamiento con sorafenib (véase la sección Efectos colaterales). Insuficiencia hepática: No hay datos disponibles de pacientes con insuficiencia hepática (grave) C de Child Pugh. Sorafenib se elimina principalmente por vía hepática, por tanto, la exposición puede aumentar en los pacientes con insuficiencia hepática grave (véase la sección Propiedades farmacocinéticas) .
|
|
Precauciones:
Embarazo y lactancia: Embarazo: No se han realizado ensayos adecuados y bien controlados en mujeres embarazadas que estuvieran tomando sorafenib. Los ensayos con animales han demostrado toxicidad en la reproducción, que incluye malformaciones (véase la sección Advertencias y Precauciones Especiales). Se demostró en ratas que sorafenib y sus metabolitos atraviesan la placenta y se supone que sorafenib inhibe la angiogénesis fetal. Durante el tratamiento, las mujeres deben evitar el embarazo. Las mujeres en edad fértil deben conocer los posibles riesgos para el feto, que incluyen malformaciones graves (teratogenia), retraso del crecimiento y muerte fetal (embriotoxicidad). Sorafenib no debe utilizarse durante el embarazo. El médico sólo debe considerar la utilización de sorafenib si los posibles beneficios justifican los riesgos potenciales para el feto (ver las secciones Advertencias y precauciones especiales, Datos preclínicos sobre seguridad). Mujeres en edad fértil: En animales, sorafenib ha demostrado ser teratógeno y embriotóxico. Durante el tratamiento y hasta al menos 2 semanas después de la terminación se utilizará un método anticonceptivo adecuado (ver las secciones Advertencias y precauciones especiales, Datos preclínicos sobre seguridad). Lactancia: No se sabe si sorafenib se excreta en la leche humana. En animales, el sorafenib y/o sus metabolitos se eliminan en la leche. Como muchos fármacos son eliminados por la leche humana y como no se han estudiado los efectos de sorafenib en la lactancia, la mujer que reciba tratamiento con sorafenib debe interrumpir la lactancia materna. Fertilidad: Los resultados de ensayos con animales indican que sorafenib puede alterar la fertilidad masculina y femenina (véase la sección Datos preclínicos sobre seguridad). Efectos sobre la capacidad para conducir o utilizar maquinaria: No se han realizado ensayos sobre los efectos de sorafenib en la capacidad de conducir o de utilizar maquinaria. No hay pruebas de que sorafenib afecte a la capacidad de conducir o de utilizar máquinas.
|
|
Interacciones Medicamentosas:
Interacciones entre medicamentos: Vía UGT1A: Se recomienda precaución si se administra sorafenib con compuestos que se metabolizan/eliminan principalmente por la vía UGT1A1 (p. ej. irinotecán) (véase la sección Interacción con otros medicamentos y otras formas de interacción). Docetaxel: El uso concomitante de docetaxel (75 ó 100 mg/m2) con sorafenib (200 ó 400 mg, 2 veces al día), administrado con una pausa posológica de 3 días en torno a la administración del docetaxel, produjo un incremento del AUC de docetaxel del 36-80%. Se recomienda precaución cuando se administre sorafenib con docetaxel (véase la sección Interacciones). Neomicina: La coadministración de neomicina puede causar una disminución de la biodisponibilidad de sorafenib (véase la sección Interacción con otros medicamentos y otras formas de interacción). Interacción con otros medicamentos y otras formas de interacción: Inductores de CYP3A4: La administración concomitante continua de sorafenib y rifampicina redujo el AUC de sorafenib en un 37%, por término medio. Otros inductores de la actividad de CYP3A4 (p. ej., Hypericum perforatum, también conocido como hierba de San Juan, fenitoína, carbamazepina, fenobarbital y dexametasona) también pueden aumentar el metabolismo de sorafenib y reducir, en consecuencia, las concentraciones de sorafenib. Inhibidores de CYP3A4: El ketoconazol, un potente inhibidor de CYP3A4, administrado 1 vez al día durante 7 días a voluntarios varones sanos no alteró el AUC media de una dosis única de 50 mg de sorafenib. Por tanto, no son probables interacciones farmacocinéticas clínicas entre sorafenib y los inhibidores de CYP3A4. Sustratos de CYP2C9: El posible efecto de sorafenib en la warfarina, un sustrato de CYP2C9, se evaluó en pacientes tratados con sorafenib, en comparación con pacientes tratados con placebo. El tratamiento concomitante con sorafenib y warfarina no dio lugar a cambios en la media de INR-TP, en comparación con el placebo. Sin embargo, es necesario controlar regularmente el INR de los pacientes que tomen warfarina (ver la sección Advertencias y precauciones especiales de empleo). Sustratos selectivos de isoformas del CYP: La administración concomitante de midazolam, dextrometorfano y omeprazol, que son sustratos de los citocromos CYP3A4, CYP2D6 y CYP2C19, respectivamente, después de 4 semanas de administración de sorafenib, no alteró la exposición de estos agentes. Esto indica que sorafenib no inhibe ni induce estas isoenzimas del citocromo P450. En un ensayo clínico distinto, la administración concomitante de sorafenib con paclitaxel produjo un aumento, en vez de una disminución, en la exposición de 6-OH paclitaxel, el metabolito activo de paclitaxel que se forma por CYP2C8. Estos datos sugieren que sorafenib puede no ser un inhibidor in vivo de CYP2C8. En otro estudio clínico, la administración concomitante de sorafenib con ciclofosfamida produjo una pequeńa disminución de la exposición a la ciclofosfamida, pero ninguna disminución de la exposición sistémica de 4-OH ciclofosfamida, el metabolito activo de la ciclofosfamida que se forma principalmente por CYP2B6. Estos datos sugieren que sorafenib puede no ser un inhibidor in vivo de CYP2B6. Combinación con otros agentes antineoplásicos: En ensayos clínicos se ha administrado sorafenib junto con una serie de agentes antineoplásicos en las pautas posológicas habituales, incluyendo gemcitabina, cisplatino, oxaliplatino, paclitaxel, carboplatino, capecitabina, doxorrubicina, docetaxel, irinotecán y ciclofosfamida. Sorafenib no tuvo efectos clínicamente relevantes en la farmacocinética de gemcitabina, cisplatino, carboplatino, oxaliplatino o ciclofosfamida. Paclitaxel/carboplatino: La administración de paclitaxel (225 mg/m2) y carboplatino (AUC = 6) con sorafenib ( Ł 400 mg 2 veces al día), administrado con una pausa posológica de 3 días en torno a la administración de paclitaxel/carboplatino, no produjo efecto significativo en la farmacocinética de paclitaxel. La coadministración de paclitaxel (225 mg/m2, 1 vez cada 3 semanas) y carboplatino (AUC=6) con sorafenib (400 mg 2 veces al día, sin pausa en la posología de sorafenib) produjo un aumento de 47% en la exposición de sorafenib, un aumento de 29% en la exposición de paclitaxel y un aumento de 50% en la exposición de 6-OH paclitaxel. La farmacocinética de carboplatino no se afectó. Estos datos indican que no es necesario un ajuste de la dosis cuando paclitaxel y carboplatino se coadministran con sorafenib con una pausa posológica de sorafenib de 3 días. Se desconoce la significación clínica de los aumentos en la exposición de sorafenib y paclitaxel en la administración conjunta de sorafenib sin una pausa posológica. Capecitabina: La coadministración de capecitabina (750-1050 mg/m2 2 veces al día, Días 1-14 cada 21 días) y sorafenib (200 ó 400 mg 2 veces al día, administración continua ininterrumpida) no produjo cambios significativos en la exposición a sorafenib, sino un aumento de 15-50% en la exposición a capecitabina y un aumento de 0-52% en la exposición a 5-FU. Se desconoce la significación clínica de estos aumentos pequeńos a moderados en la exposición a capecitabina y 5-FU cuando se coadministraron con sorafenib. Doxorrubicina/irinotecán: El tratamiento concomitante con sorafenib produjo un incremento del 21% del AUC de doxorrubicina. Cuando se administró con irinotecán, cuyo metabolito activo SN-38 se sigue metabolizando por la vía UGT1A1, se produjo un incremento del 67-120% del AUC de SN-38 y un incremento del 26-42% del AUC de irinotecán. No se conoce la relevancia clínica de estos resultados (véase la sección Advertencias y precauciones especiales de empleo). Docetaxel: El docetaxel (75 ó 100 mg/m2, administrados 1 vez cada 21 días) coadministrado con sorafenib (200 mg, 2 veces al día, o 400 mg, 2 veces al día, administrados desde el día 2 al 19 de un ciclo de 21 días) con una pausa posológica de 3 días en torno a la administración de docetaxel, produjo un aumento del 36-80% en el AUC de docetaxel y del 16-32% en la Cmáx de docetaxel. Se aconseja precaución cuando se administre sorafenib con docetaxel (ver la sección Advertencias y precauciones especiales de empleo). Combinación con otros antibióticos: Neomicina. La coadministración de neomicina, un agente antimicrobiano no sistémico usado para erradicar flora GI, interfiere la circulación enterohepática de sorafenib (ver Propiedades farmacocinéticas, Metabolismo y eliminación), ocasionando una disminución de la exposición a sorafenib. En voluntarios sanos tratados con un régimen de 5 días de neomicina, la biodisponibilidad promedio de sorafenib disminuyó un 54%. Se desconoce la relevancia clínica de estos hallazgos. No se han estudiado los efectos de otros antibióticos, pero dependerán probablemente de su capacidad para disminuir la actividad de la glucuronidasa. Combinación con inhibidores de la bomba de protones: Omeprazol: La administración concomitante de omeprazol no afecta la farmacocinética de sorafenib. No es necesario ajustar la dosis de sorafenib.
|
|
Sobredosificación:
No hay ningún tratamiento específico para la sobredosis de sorafenib. La dosis mayor de sorafenib estudiada clínicamente es de 800 mg, 2 veces al día. Las reacciones adversas observadas a esta dosis eran principalmente diarrea y eventos dermatológicos. Si se sospecha una sobredosis, se interrumpirá la administración de sorafenib y se establecerá tratamiento de apoyo.
|
|
Observaciones:
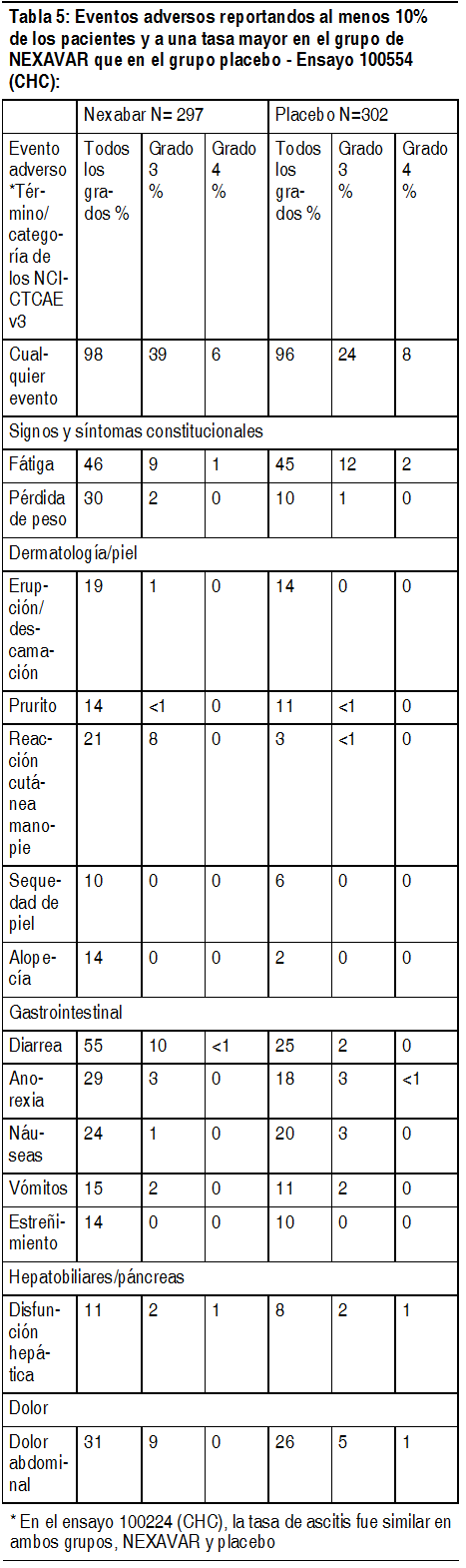
Apéndices: Apéndice 1: Presentación en los EE. UU. PI: Las reacciones adversas más comunes fueron diarrea, erupción, alopecia y síndrome de mano y pie (corresponde al síndrome de eritrodisestesia palmoplantar del MedDRA). Los datos descritos en estas secciones reflejan la exposición a Nexavar de 748 pacientes que participaron en ensayos controlados con placebo en carcinoma hepatocelular (n=297) o carcinoma de células renales avanzado (n=451). Las reacciones adversas más frecuentes ( ł 20%), que en los pacientes con CHC o CCR se consideraron relacionadas con Nexavar, son fatiga, pérdida de peso, erupción/descamación, reacción cutánea en manos y pies, alopecia, diarrea, anorexia, náuseas y dolor abdominal. Como los ensayos clínicos se llevaron a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no se pueden comparar directamente con las tasas en los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica. Reacciones adversas en el estudio sobre el carcinoma hepatocelular (CHC) 100554: La Tabla 5 muestra el porcentaje de pacientes con CHC que experimentaron reacciones adversas informadas en al menos 10% de los pacientes y a una tasa más alta en el grupo de Nexavar que en el de placebo en el Estudio 100554. Se informó de reacciones adversas de grado 3 de los CTCAE en 39% de los pacientes que recibieron Nexavar, en comparación con 24% de los que recibieron placebo. Si informó de eventos adversos de grado 4 de los CTCAE en 6% de los pacientes que recibieron Nexavar, en comparación con 8% de los que recibieron placebo.
Ver Tabla
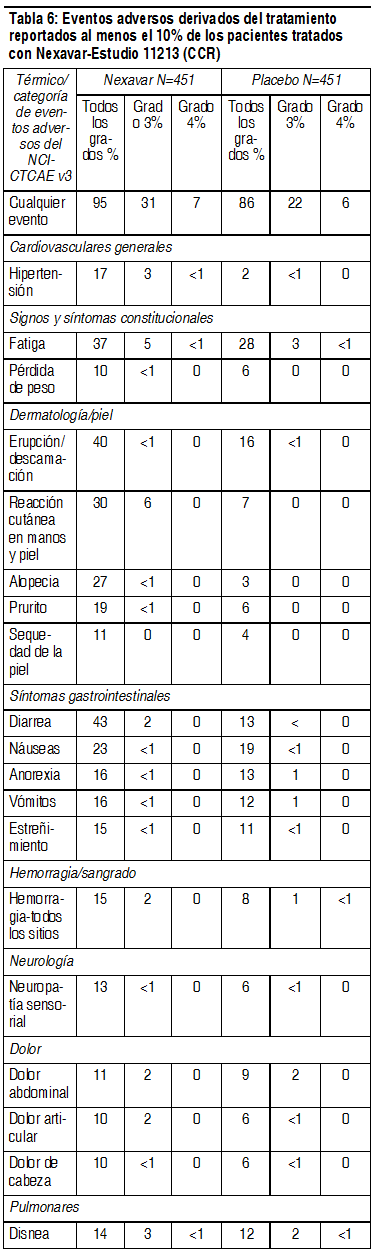
Se reportó hipertensión en 9% de los pacientes tratados con Nexavar y en 4% de los tratados con placebo. Se reportó hipertensión de grado 3 de CTCAE en 4% de los pacientes tratados con Nexavar y en 1% de los tratados con placebo. No se reportó que algún paciente tuviera eventos de grado 4 de CTCAE en ningún grupo de tratamiento. Se reportó hemorragia/sangrado en 20% de los pacientes tratados con placebo y en 18% de los tratados con Nexavar. Las tasas de sangrado de grados 3 y 4 de CTCAE también fueron mayores en el grupo tratado con placebo (grado 3 de CTCAE en 5% del grupo de placebo y 3% del grupo de Nexavar, y grado 4 de CTCAE en 4% del grupo de placebo y 2% del grupo de Nexavar). Se reportó sangrado de várices esofágicas en 4% de los pacientes tratados con placebo y 2.4% de los tratados con Nexavar. Se reportó insuficiencia renal en 2.6% de los pacientes tratados con placebo y en 0.3% de los tratados con Nexavar. Reacciones adversas en el estudio 11213 sobre CCR: En la Tabla 6 se presenta el porcentaje de pacientes con CCR que tuvieron eventos adversos reportados al menos por 10% de los pacientes y a una tasa mayor en el grupo de Nexavar que en el grupo de placebo, en el estudio 11213. Se informó de eventos adversos de grado 3 de los CTCAE en 31% de los pacientes que recibieron Nexavar, en comparación con 22% de los que recibieron placebo. Se informó de eventos adversos de grado 4 de los CTCAE en 7% de los pacientes que recibieron Nexavar, en comparación con 6% de los que recibieron placebo.
Ver Tabla
Datos adicionales de ensayos clínicos múltiples: Los siguientes eventos adversos y anomalías de laboratorio adicionales relacionados con el fármaco fueron reportados en ensayos clínicos de sorafenib (muy frecuentes: 10% o más; frecuentes: de 1% a menos de 10%; poco frecuentes: de 0.1% a menos de 1%; raros: de 0.01% a menos de 0.1%). Cardiovasculares: Frecuentes: insuficiencia cardíaca congestiva*; isquemia y/o infarto del miocardio* Poco frecuentes: crisis hipertensiva* Raros: prolongación del intervalo QT. Insuficiencia cardíaca congestiva: En ensayos clínicos patrocinados por la compańía, la insuficiencia cardíaca congestiva se reportó como evento adverso en 1.9% de los pacientes tratados con sorafenib (N=2276). En el estudio 11213 (CCR) se tuvieron informes de eventos adversos congruentes con la insuficiencia cardíaca congestiva en 1.7% de los pacientes tratados con sorafenib y 0.7% de los tratados con placebo. En el estudio 100554 (CHC), se informó de estos eventos en 0.99% de los pacientes tratados con sorafenib y 1.1% de los tratados con placebo. Dermatológicos: Muy frecuentes: eritema; Frecuentes: dermatitis exfoliativa, acné, rubefacción. Poco frecuentes: foliculitis, eccema, eritema multiforme, queratoacantomas/ carcinoma de células escamosas de la piel, síndrome de Stevens- Johnson. Digestivos: Muy frecuentes: aumento de la lipasa, aumento de la amilasa; Frecuentes: mucositis, estomatitis (incluyendo sequedad de boca y glosodinia), dispepsia, disfagia; Poco frecuentes: pancreatitis, reflujo gastrointestinal, gastritis, perforaciones gastrointestinales*. Nótese que las elevaciones de la lipasa son muy frecuentes (41%, véase más adelante); el diagnóstico de pancreatitis no se debe establecer solamente en base a cifras anormales de laboratorio. Trastornos generales: Muy frecuentes: hemorragia (incluyendo los tractos gastrointestinal* y respiratorio* y casos poco frecuentes de hemorragia cerebral*), astenia, dolor (incluyendo dolores de boca, huesos y tumores). Frecuentes: disminución del apetito, enfermedad pseudogripal, pirexia. Poco frecuentes: infección. Hematológicos: Muy frecuentes: leucopenia, linfopenia. Frecuentes: anemia, neutropenia, trombocitopenia. Poco frecuentes: INR anormal. Hipersensibilidad: Poco frecuentes: reacciones de hipersensibilidad (incluyendo reacciones cutáneas y urticaria). Metabólicos y nutricionales: Muy frecuentes: hipofosfatemia. Frecuentes: aumentos transitorios de las transaminasas. Poco frecuentes: deshidratación, hiponatremia, aumentos transitorios de la fosfatasa alcalina, aumento de la bilirrubina (incluyendo ictericia), hipotiroidismo, hipertiroidismo, colecistitis, colangitis. Musculoesqueléticos: Frecuentes: artralgia, mialgia. Sistema nervioso y psiquiátricos: Frecuentes: depresión. Poco frecuentes: acúfenos, leucoencefalopatía posterior reversible*. Renales/Genitourinarios: Frecuentes: insuficiencia renal. Reproductores: Frecuentes: disfunción eréctil; Poco frecuentes: ginecomastia. Respiratorios: Frecuentes: ronquera. Poco frecuentes: rinorrea, eventos similares a la enfermedad pulmonar intersticial (incluye informes de neumonitis, neumonía por radiación, angustia respiratoria aguda, neumonía intersticial, neumonitis e inflamación pulmonar) *eventos que pueden hacer peligrar la vida o tener un desenlace mortal. Tales eventos son poco frecuentes o incluso más raros. Además, los siguientes eventos adversos médicamente significativos se reportaron con poca frecuencia durante ensayos clínicos de Nexavar: accidente isquémico transitorio, arritmia y tromboembolismo. No se estableció la relación causal entre estos eventos y Nexavar. Dos ensayos clínicos aleatorizados y controlados con placebo que compararon la seguridad y eficacia de sorafenib en combinación con quimioterapias dobles basadas en el platino (carboplatino/paclitaxel y separadamente, gemcitabina/cisplatino) frente a las respectivas quimioterapias dobles basadas en el platino, a solas, como tratamiento de primera línea en pacientes con carcinoma pulmonar de células no pequeńas (NSCLC) en estado avanzado no cumplió su criterio de valoración primario de mejorar la supervivencia global. Los eventos de seguridad fueron generalmente congruentes con los reportados previamente. Sin embargo, en ambos ensayos se observó mayor mortalidad en el subgrupo de pacientes con carcinoma pulmonar de células escamosas tratado con sorafenib y quimioterapias dobles basadas en el platino, frente a los tratados con quimioterapias dobles basadas en el platino, a solas (carboplatino/paclitaxel: IR 1.81, CI de 95% 1.19-2,74; gemcitabina/cisplatino: IR 1.22, CI de 95% 0.82-1.80). No se identificó ninguna causa definitiva de los hallazgos. La seguridad también se evaluó en un conjunto de estudios de fase 2, constituido por 638 pacientes tratados con Nexavar, 202 de ellos con CCR, 137 con carcinoma hepatocelular y 299 con otros tipos de cáncer. Los eventos adversos más frecuentes relacionados con el fármaco que se reportaron en los pacientes tratados con sorafenib en este conjunto, fueron erupción (38%), diarrea (37%), reacción cutánea de manos y pies (35%) y fatiga (33%). Las tasas respectivas de eventos adversos relacionados con el medicamento de grado 3 y 4 de los CTC (v 2.0) en los pacientes tratados con Nexavar fueron de 37% y 3%, respectivamente. Experiencias de eventos adversos posteriores a la comercialización: Se han identificado las siguientes reacciones farmacológicas adversas durante el uso posterior a la autorización de Nexavar. Debido a que estas reacciones se reportan voluntariamente en una población de tamańo incierto, no siempre es posible estimar de forma confiable su frecuencia, o establecer una relación causal con la exposición al fármaco. Dermatológicos: dermatitis por recuerdo de radiación, síndrome de Stevens-Johnson, vasculitis leucocitoclástica. Hipersensibilidad: angioedema, reacción anafiláctica. Trastornos hepatobiliares: Hepatitis inducida por el fármaco (se han observado casos potencialmente mortales y fatales). Musculoesqueléticos: rabdomiólisis.
|
|
Presentaciones:
Envase conteniendo 112 comprimidos.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}