 |
Composición:
Cada comprimido recubierto contiene: Lapatinib Ditosilato Monohidratado, equivalente a 250 mg de Lapatinib. Lista de excipientes: Núcleo: Almidón Glicolato de Sodio, Estearato de Magnesio, Celulosa Microcristalina, Povidona K-30. Recubrimiento: Hipromelosa, Dióxido de Titanio, Macrogol 400, Polisorbato 80, Oxido de Hierro Amarillo, Oxido de Hierro Rojo. Los comprimidos de 250 mg de lapatinib (como ditosilato) son ovalados y biconvexos, tienen una película de recubrimiento amarillo y uno de sus lados es liso, mientras que el opuesto se encuentra grabado con las siglas GS XJG. Presentación farmacéutica: Envase conteniendo 70 comprimidos recubiertos.
|
|
Indicaciones:
Tykerb®, en combinación con capecitabina, se indica en el tratamiento de pacientes que padecen cáncer de mama avanzado o metastásico, cuyos tumores sobreexpresan la proteína ErbB2 (HER2/neu), y que han recibido tratamiento previo incluyendo una antraciclina, un taxano y trastuzumab (véase Estudios Clínicos). Tykerb®, en combinación con letrozol, está indicado en el tratamiento de pacientes postmenopáusicas con cáncer de mama avanzado o metastásico, con receptores hormonales positivos y cuyos tumores sobreexpresan la proteína ErbB2 (HER2/neu) (véase Estudios Clínicos).
|
|
Propiedades:
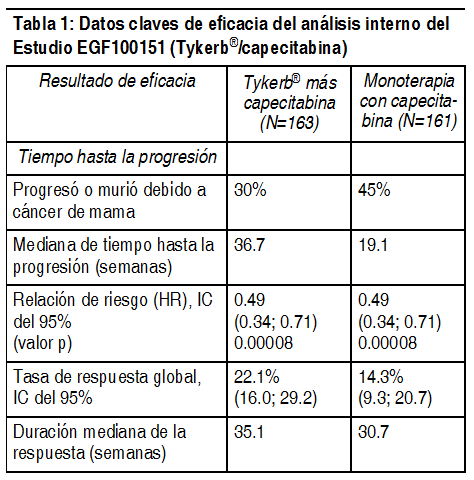
Farmacodinamia: Mecanismo de acción: El lapatinib es un novel inhibidor de la 4-anilinoquinazolin cinasa, con un mecanismo único de acción, ya que es un potente inhibidor selectivo y reversible de los dominios intracelulares de tirosina cinasa, tanto de los receptores ErbB1 como del ErbB2 (HER2) (valores Kiapp estimados de 3nM y 13nM, respectivamente), con una velocidad lenta de disociación de estos receptores (vida media mayor o igual a 300 minutos). Se descubrió que esta velocidad de disociación es menor que la de los otros inhibidores de la 4-anilinoquinazolin cinasa estudiados. El lapatinib inhibe el crecimiento celular tumoral dirigido por ErbB, tanto in vitro como en diversos modelos animales. Además de su actividad como agente único, se demostró que el lapatinib ejerce un efecto aditivo en un estudio realizado in vitro, en el cual se utilizó la combinación de lapatinib y 5-FU (el metabolito activo de la capecitabina) en las 4 líneas celulares tumorales analizadas. Se desconoce el significado clínico de esta información obtenida in vitro. En líneas celulares acondicionadas con trastuzumab, se evaluaron los efectos inhibidores de crecimiento que ejerce el lapatinib. El lapatinib mantuvo un nivel significativo de actividad contra las líneas celulares de cáncer de mama seleccionadas para un crecimiento a largo plazo en un medio in vitro que contenía trastuzumab. Estos hallazgos sugieren la ausencia de resistencia cruzada entre estos 2 agentes directos ErbB2. Las células del cáncer de mama sensibles a hormonas (receptores de estrógenos [ER] y/o receptores de progesterona [PgR]) positivos, que coexpresan la proteína ErbB2, tienden a volverse resistentes a las terapias endocrinas establecidas. Las células del cáncer de mama sensibles a hormonas que carecen inicialmente de ErbB1 o ErbB2 regularían ascendentemente estos receptores conforme el tumor se va volviendo resistente a la terapia endocrina. Estudios aleatorizados que se realizaron en pacientes con cáncer de mama metastásico y sensible a hormonas indicaron que un inhibidor ErbB2 o ErbB1 de la tirosina cinasa podría mejorar potencialmente el perfil de eficacia clínica cuando se adiciona a una terapia endocrina. Farmacocinética: Absorción: La absorción posterior a la administración oral de lapatinib es incompleta y variable (con un coeficiente de variación de aproximadamente 50 a 100% en el ABC). Las concentraciones séricas se hacen presentes una vez que transcurre un lapso de tiempo mediano de 0.25 horas (intervalo de 0 a 1.5 horas). Las concentraciones plasmáticas máximas (Cmáx) de lapatinib se alcanzan aproximadamente 4 horas después de su administración. La dosificación diaria de 1250 mg produce valores geométricos promedio de Cmáx en estado estacionario (intervalo de confianza del 95%) de 2.43 (1.57 a 3.77) µg/ml, así como valores del Área Bajo la Curva (ABC) de 36.2 (23.4 a 56) µg*hr/mL. La exposición sistémica al lapatinib es mayor cuando el fármaco se administra junto con alimentos (ver Posología e Interacciones). Los valores de ABC del lapatinib fueron aproximadamente 3 y 4 veces mayores (Cmáx aproximadamente 2.5 y 3 veces mayor), cuando el fármaco se administró con una comida baja en grasa (5% de grasa [500 calorías]) o con una comida alta en grasa (50% de grasa [1.000 calorías]), respectivamente. Distribución: El lapatinib exhibe un alto grado de fijación proteínico (superior al 99%) a la albúmina y a la alfa-1-glucoproteína ácida. Los estudios realizados in vitro indican que el lapatinib es un sustrato de los transportadores de la proteína resistente al cáncer de mama (BCRP, por sus siglas en inglés; ABCG1) y la p-glucoproteína (ABCB1). También se ha demostrado que el lapatinib inhibe Pgp (IC50 2,3 µg/mL), BCRP (IC50 0,014 µg/mL) y, así como el transportador de captación hepática OATP 1B1(IC50 2,3 µg/mL), in vitro a concentraciones clínicamente importantes. Se desconoce la importancia clínica de estos efectos en el perfil farmacocinético de otros fármacos, o en la actividad farmacológica de otros agentes anticancerígenos. El lapatinib no inhibe significativamente a los transportadores renales OAT o OCT (los valores in vitro de IC50 fueron mayores o iguales a 6,9 µg/mL). Metabolismo: El lapatinib experimenta un extenso metabolismo, el cual tiene lugar primariamente a través de CYP3A4 y CYP3A5, con una contribución menor de las enzimas CYP2C19 y CYP2C8 hacia una variedad de metabolitos oxidados, de los cuales ninguno representa más del 14% de la dosis recuperada en las heces, ni más del 10% de la concentración plasmática de lapatinib. A concentraciones clínicamente importantes, el lapatinib inhibe las enzimas CYP3A (Ki de 0.6 a 2.3 µg/ml) y CYP2C8 (0.3 µg/ml) in vitro. En los microsomas hepáticos humanos, el lapatinib no inhibió significativamente las siguientes enzimas: CYP1A2, CYP2C9, CYP2C19 y CYP2D6, ni las enzimas UGT (los valores in vitro de CI50 fueron mayores o iguales a 6.9 µg/ml). En voluntarios sanos que recibieron ketoconazol, un inhibidor de la enzima CYP3A4, en un régimen de dosificación de 200 mg administrados 2 veces al día durante 7 días, la exposición sistémica al lapatinib aumentó 3.6 veces, mientras la vida media aumentó 1.7 veces. En voluntarios sanos que recibieron carbamazepina, un inductor de la enzima CYP3A4, en un régimen de dosificación de 100 mg administrados 2 veces al día durante 3 días y 200 mg administrados 2 veces al día durante 17 días, la exposición sistémica al lapatinib disminuyó aproximadamente un 72%. Eliminación: La vida media del lapatinib, cuantificada después de administrar dosis únicas, aumentó de manera proporcional con los incrementos de las dosis. Sin embargo, la dosificación diaria de lapatinib permite alcanzar el estado estacionario o de equilibrio en un lapso de 6 a 7 días, lo cual indica que la vida media eficaz es de 24 horas. El lapatinib se elimina principalmente por metabolismo de CYP3A4/5. La ruta primaria de eliminación del lapatinib y sus metabolitos son las heces, donde menos del 2% de la dosis (como lapatinib y metabolitos) se excreta en la orina. La porción de la dosis de lapatinib que se recupera en las heces representa una mediana de 27% (intervalo de 3 a 67%) de una dosis oral. Poblaciones de pacientes especiales: Insuficiencia renal: No se han realizado estudios específicos para evaluar el perfil farmacocinético del lapatinib en pacientes que padecen insuficiencia renal, ni en pacientes sometidas a hemodiálisis. Sin embargo, es improbable que la presencia de insuficiencia renal afecte el perfil farmacocinético del lapatinib, debido a que menos del 2% de una dosis administrada se elimina por la vía renal (como lapatinib inalterado y metabolitos). Insuficiencia hepática: Se examinó el perfil farmacocinético del lapatinib en pacientes con insuficiencia hepática de grado moderado (n = 8) o severo (n = 4) (puntuación de Child-Pugh de 7-9, o más de 9, respectivamente), y en 8 sujetos control sanos. Después de administrar una dosis oral única de 100 mg, la exposición sistémica (ABC) al lapatinib aumentó aproximadamente 56% y 85%, en pacientes con insuficiencia hepática de grado moderado y severo, respectivamente. Se deberá proceder con precaución al administrar lapatinib a pacientes que padezcan insuficiencia hepática (ver Posología y Advertencias y Precauciones). Estudios clínicos: Tykerb® en combinación con capecitabine: En un estudio aleatorio de Fase III, se evaluó la seguridad y la eficacia de Tykerb®, administrado en combinación con capecitabina en el tratamiento del cáncer de mama. Las pacientes elegibles para participar en el estudio tenían el diagnóstico de cáncer de mama metastásico o localmente avanzado, que sobre-expresaban la proteína ErbB2, y que habían progresado después de tratamiento previo que incluyo taxanos, antraciclinas y trastuzumab. Antes de iniciar el tratamiento con Tykerb®, se evaluó la FEVI en todas las pacientes (utilizando un ecocardiograma o MUGA) para garantizar que la FEVI basal se encontrara dentro de los límites institucionales normales. En los estudios clínicos, durante todo el tratamiento con lapatinib, se vigiló la FEVI a intervalos de aproximadamente, cada 8 semanas para garantizar que no disminuyera por debajo del límite institucional inferior normal. La mayoría de las disminuciones en la FEVI (superiores al 60%) se produjeron durante las primeras nueve semanas de tratamiento. Sin embargo, existe poca información sobre la exposición a largo plazo. Las pacientes fueron distribuidas aleatoriamente para recibir ya sea 1250 mg de Tykerb®, administrados 1 vez al día (de manera continua), más capecitabina (2000 mg/m2/día, los días 1-14, cada 21 días), o bien, para recibir una monoterapia con capecitabina (2500 mg/m2/día, los días 1-14, cada 21 días). En el estudio el tratamiento fue administrado hasta la progresión de la enfermedad, o retiro del estudio por alguna razón. El objetivo primario de valoración fue el tiempo hasta la progresión (TTP); los resultados presentados más adelantes están basados en la revisión llevada a cabo por una comisión o panel de revisión independiente. Se realizó un análisis interino preespecificado con una fecha de corte de datos del 15 de noviembre de 2005. Este análisis demostró una mejoría en el TTP (reducción del 51% en el riesgo de desarrollar progresión) para las pacientes que se encontraban recibiendo tratamiento con Tykerb® más Capecitabina, en comparación con capecitabina sola.
Ver Tabla
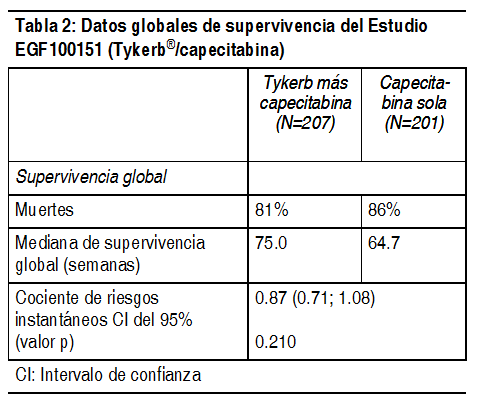
Se realizó un análisis subsiguiente con una fecha de corte de datos del 03 de abril de 2006 (la fecha en la que se detuvo el reclutamiento ulterior). En ese momento, se había reclutado a un total de 399 pacientes (198 para el grupo de combinación, 201 para el grupo de control). El análisis realizado por un panel independiente de revisión confirmó una mejoría en el TTP, con una reducción de 43% en el riesgo de desarrollar la progresión en pacientes que recibían Tykerb más capecitabina, en comparación con capecitabina sola (p=0.00013). La mediana del TTP fue de 27.1 y 18.6 semanas, la respuesta global fue de 23.7% y 13.9% y la mediana de la duración de la respuesta fue de 32.1 y 30.6 semanas, para los grupos tratados con Tykerb más capecitabina y capecitabina sola, respectivamente. En el grupo que recibió la terapia de combinación hubo 4 (2%) progresiones en el sistema nervioso central, en comparación con las 13 (6%) progresiones observadas en el grupo que recibió la monoterapia con capecitabina. de acuerdo a la evaluación de un panel independiente de revisión. En el momento en que se detuvo el reclutamiento para el estudio EGF100151 (03 de abril de 2006), 399 pacientes fueron aleatorizados a la terapia del estudio, mientras que otros 9 estaban siendo evaluados para ser seleccionados. A los 9 pacientes que se encontraban en el período de selección, así como a todos aquellos que ya se encontraban recibiendo monoterapia con capecitabina, se les ofreció el tratamiento con combinación (capecitabina con lapatinib). En total, 207 pacientes fueron asignados a la terapia de combinación y 201 pacientes fueron asignados a la monoterapia con capecitabina. En la Tabla 2 se resumen un análisis de datos de supervivencia al 01 de octubre de 2008.
Ver Tabla
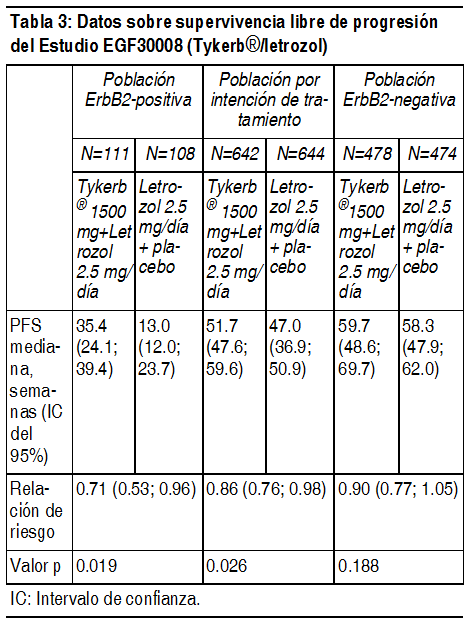
Después de detenido el enrolamiento en el estudio, se transfirió a 36 pacientes del grupo de capecitabina al grupo tratado con lapatinib + capecitabina, de los cuales 26 fueron transferidos antes de progresión de la enfermedad mientras se encontraban recibiendo capecitabina sola. Para poder aislar el efecto terapéutico en la presencia de la transferencia, se realizó un análisis de regresión de Cox considerando la transferencia como una covariable dependiente del tiempo y el efecto del tratamiento. Los resultados de este análisis sugieren una reducción clínicamente relevante en el riesgo de muerte de 20%, con un cociente de riesgos instantáneos en el efecto terapéutico de 0,80 (intervalo de confianza de 95% [CI]: 0.64; 0.99; p=0.043). Tratamiento de combinación con Tykerb ® y letrozol: Tykerb ha sido estudiado en combinación con letrozol para el tratamiento del cáncer de mama avanzado o metastásico, en mujeres postmenopáusicas con receptores hormonales positivos (receptores de estrógenos [ER] y/ receptores de progesterona [PgR] positivos). EGF30008 fue un estudio aleatorio, doble ciego y controlado en pacientes con cáncer de mama localmente avanzado o metastásico (MBC, por sus siglas en inglés), con receptores hormonales positivos (HR+), que no habían recibido previamente, terapia sistémica alguna para el tratamiento de su enfermedad metastásica. 1286 pacientes fueron distribuidos aleatoriamente para recibir 2.5 mg de letrozol, 1 vez al día, más 1.500 mg de Tykerb 1 vez al día o letrozol con placebo. La distribución aleatoria fue estratificada por sitios de enfermedad y terapia previa con antiestrógenos adyuvantes. Se determinó retrospectivamente el estado del receptor ErbB2 mediante pruebas en el laboratorio central. De todas las pacientes distribuidas aleatoriamente a tratamiento, 219 tuvieron tumores que sobreexpresaron el receptor ErbB2 (la 'población ErbB2-positiva'), la cual constituyó la población primaria preespecificada para el análisis de eficacia. Hubo 952 pacientes ErbB2-negativos y un total de 115 pacientes cuyo estado de ErbB2 no fue confirmado. En la población de pacientes ErbB2-positivos, la supervivencia libre de progresión (PFS, por sus siglas en inglés) determinada por el investigador fue significativamente mayor con la combinación de letrozol más Tykerb que con la combinación de letrozol más placebo (véase Tabla 2).
Ver Tabla
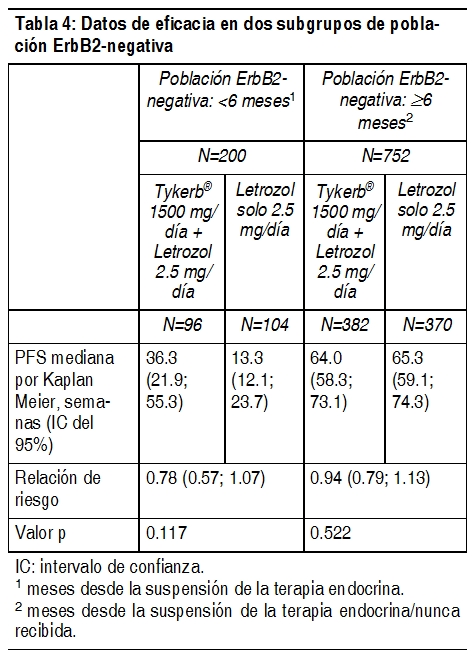
Ver Tabla
Información preclínica de seguridad: Se realizaron estudios de Tykerb en ratonas y conejas preńadas que recibieron dosis orales de 30, 60 y 120 mg/kg/día. No se observaron efectos teratogénicos; sin embargo, se produjeron anomalías menores (arteria umbilical hacia el lado izquierdo, costillas cervicales y osificación precoz) en ratas que recibieron dosis maternas tóxicas de 120 mg/kg/día (6,4 veces la exposición clínica esperada en humanos administrando 1.250 mg de lapatinib y 2000 mg/m2 de capecitabina). En las conejas, Tykerb estuvo asociado con toxicidad materna a dosis de 60 y 120 mg/kg/día (6,5% y 19% de la exposición clínica esperada en humanos administrando 1250 mg de lapatinib y 2000 mg/m2 de capecitabine, respectivamente) y abortos a dosis de 120 mg/kg/día. La toxicidad materna estuvo asociada con una disminución en los pesos corporales de los fetos, y con variaciones esqueléticas menores. En el estudio de desarrollo prenatal y postnatal realizado en ratas, se produjo una disminución en la tasa de supervivencia de las crías, entre el nacimiento y el día 21 postnatal, a dosis de 60 mg/kg/día o mayores (3.3 veces la exposición clínica esperada en humanos administrando 1250 mg de lapatinib y 2000 mg/m2 de capecitabina). La dosis más alta que no produjo efecto alguno en este estudio fue de 20 mg/kg/día. En estudios de carcinogenicidad oral realizados con lapatinib, se observaron lesiones cutáneas severas a las dosis más altas analizadas, las cuales produjeron niveles de exposición basados en ABC de hasta 1.7 veces, en ratones y ratas machos, y de hasta 12 veces en ratas hembras, en comparación con los observados en seres humanos administrando 1.250 mg de lapatinib y 2.000 mg/m2 de capecitabina. No hubo indicios de carcinogenicidad en ratones. En ratas, la incidencia de hemangioma benigno de los nódulos linfáticos mesentéricos fue mayor en algunos grupos que en los controles concurrentes, pero estuvo dentro del rango de referencia. También se observó un incremento en la incidencia de infartos renales y necrosis papilares en ratas hembras, a niveles de exposición 6 y 8 veces mayores que los observados en seres humanos administrando 1.250 mg de lapatinib y 2.000 mg/m2 de capecitabina. Es incierta la relevancia de estos hallazgos para los seres humanos. Tykerb no fue clastogénico ni mutagénico en un conjunto de ensayos que incluyeron el ensayo de aberración cromosómica en hámsteres chinos, el ensayo de Ames, el ensayo de aberración cromosómica en linfocitos humanos y un ensayo de aberración cromosómica in vivo en la médula ósea de ratas. No se observaron efectos en la fertilidad, apareamiento o función gonadal de ratas machos o hembras, a dosis de hasta 120 mg/kg/día (hembras) y de hasta 180 mg/kg/día (machos) (8 y 3 veces la exposición clínica esperada en los seres humanos, respectivamente). Se desconoce el efecto que ejerce Tykerb en la fertilidad humana.
|
|
Posología:
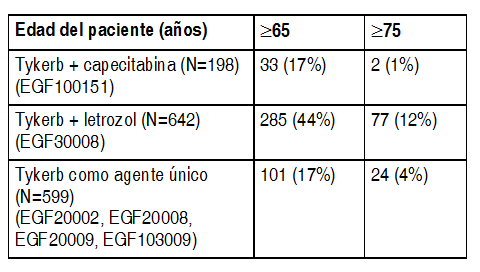
Sólo un médico con experiencia en la administración de agentes anticancerígenos deberá iniciar el tratamiento con Tykerb®. Antes de iniciar el tratamiento, se deberá evaluar la fracción de eyección del ventrículo izquierdo (FEVI) para garantizar que la FEVI basal se encuentre dentro de los límites institucionales normales (ver Advertencias y Precauciones). Durante todo el tratamiento con Tykerb®, se deberá seguir vigilando la FEVI para garantizar que no disminuya por debajo del límite institucional inferior normal (véase Demora y reducción en la dosificación - eventos cardíacos). Tykerb® debe tomarse cuando menos 1 hora antes o 1 hora después de los alimentos (ver Interacciones y Farmacocinética - Absorción). No se deben reemplazar las dosis omitidas, sino reiniciar la dosificación con la siguiente dosis diaria programada (ver Posología). Consulte la información completa para prescribir del medicamento coadministrado, para conocer los detalles pertinentes sobre su posología, contraindicaciones e información de seguridad. Tykerb® en combinación con capecitabina: La dosis recomendada de Tykerb® consiste en 1250 mg (es decir, 5 comprimidos) administrados 1 vez al día de manera continua cuando se toma en combinación con capecitabina. La dosis recomendada de capecitabina consiste en 2000 mg/m2/día, tomados en 2 dosis administradas cada 12 horas, los días 1-14 de un ciclo de 21 días (ver Estudios Clínicos). La capecitabina debe administrarse acompańada de alimentos, o bien, dentro de los 30 minutos posteriores a la ingestión de los mismos. Tykerb® en combinación con Letrozol: La dosis recomendada de Tykerb® consiste en 1500 mg (es decir, 6 tabletas), 1 vez al día de manera continua, cuando se toma en combinación con letrozol. Cuando Tykerb® se coadministra con el inhibidor de la aromatasa, letrozol, la dosis recomendada de letrozol consiste en 2.5 mg 1 vez al día. Retraso y reducción en la dosificación: Eventos cardíacos (ver Advertencias y Precauciones): Se deberá suspender el tratamiento con Tykerb® en aquellas pacientes que experimenten síntomas asociados con una disminución en la FEVI, de grado 2 o mayor según los Criterios Terminológicos Comunes para Eventos Adversos del Instituto Nacional de Cancerología (CTCEA del NCI), o si su FEVI disminuye por debajo del límite inferior institucional normal. Una vez que hayan transcurrido cuando menos 2 semanas, y la FEVI se haya normalizado y la paciente se encuentre asintomática, se podrá reiniciar el tratamiento con Tykerb® a dosis reducidas (1000 mg/día cuando se administre con capacitabina ó 1250 mg al día cuando se administre con letrozol). Según la información disponible actualmente, la mayoría de las disminuciones en la FEVI se producen durante las primeras 9 semanas de tratamiento. Sin embargo, existe poca información sobre la exposición a largo plazo. Enfermedad pulmonar intersticial/ neumonitis (ver Advertencias y Precauciones y Efectos colaterales): Se deberá descontinuar el tratamiento con Tykerb® en pacientes que experimenten síntomas pulmonares indicativos de enfermedad pulmonar intersticial/neumonitis que sean grado 3 o superior de acuerdo al CTCEA del NCI. Otras toxicidades: Cuando una paciente desarrolla una toxicidad de grado 2 o superior, según los CTCEA del NCI, se podría contemplar la interrupción o descontinuación del tratamiento con Tykerb®. Existe la posibilidad de restablecer la dosificación a 1250 mg/día cuando se administra con capacitabina o bien, 1500 mg/día cuando se administra con letrozol, cuando la toxicidad mejora a grado 1 o menos. Si se produce una recurrencia de toxicidad, entonces la terapia con Tykerb® deberá reiniciarse a una dosis menor (1000 mg/día cuando se administra con capecitabina, ó 1250 mg/día cuando se administra con letrozol). Poblaciones: Insuficiencia renal: No se cuenta con experiencia en la administración de Tykerb® a pacientes con insuficiencia renal severa. Sin embargo, es improbable que las pacientes que padezcan insuficiencia renal requieran una modificación en su dosificación de Tykerb®, debido a que menos del 2% de la dosis administrada (lapatinib y sus metabolitos) se elimina por la vía renal (ver Farmacocinética - Poblaciones de Pacientes Especiales). Insuficiencia hepática: El lapatinib es metabolizado a nivel hepático. La insuficiencia hepática de grado moderado y severo ha sido asociada con aumentos del 56% y 85% en la exposición sistémica, respectivamente. Se deberá tener precaución al administrar la terapia con Tykerb® a pacientes que padezcan insuficiencia hepática, debido a que este grupo de pacientes experimenta un mayor grado de exposición al fármaco (ver Advertencias y Precauciones y Farmacocinética - Poblaciones de Pacientes Especiales). Se debe reducir la dosis de Tykerb® en pacientes con insuficiencia hepática severa (Clase C de Child-Pugh). Se predice una reducción posológica de 1250 mg a 750 mg/día, o de 1500 mg/día a 1000 mg/día, en pacientes con insuficiencia hepática severa para ajustar el área bajo la curva (ABC) al rango normal. Sin embargo, no existen datos clínicos con este ajuste posológico en pacientes con insuficiencia hepática severa (ver Advertencias y Precauciones y Farmacocinética - Poblaciones de Pacientes Especiales). Nińos: Aún no se establecen la seguridad y la eficacia de Tykerb® en pacientes pediátricos. Pacientes de edad avanzada: Existe poca información concerniente al uso de Tykerb® en pacientes de 65 ańos de edad y mayores.
Ver Tabla
Con base en la edad, no se observaron diferencias generales en la seguridad y eficacia de estos regímenes. En otra experiencia clínica reportada no se han identificado diferencias en las respuestas entre pacientes de edad avanzada y pacientes más jóvenes. No es posible descartar una mayor sensibilidad de los individuos de edad avanzada.
|
|
Efectos Colaterales:
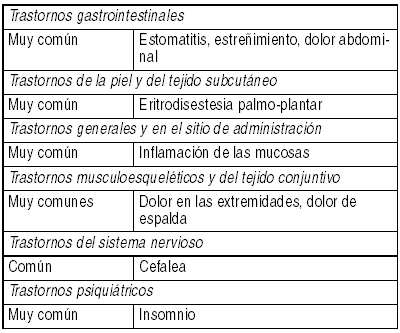
Información obtenida a partir de estudios clínicos: La seguridad de Tykerb® ha sido evaluada en más de 11.000 pacientes, tanto en estudios de monoterapia, como en estudios de combinación con otros agentes quimioterapéuticos indicados para diversos tipos de cáncer, incluyendo 198 pacientes que recibieron Tykerb® en combinación con capecitabina y 654 pacientes que recibieron Tykerb® en combinación con letrozol (ver Estudios Clínicos). Se ha utilizado la siguiente convención para la clasificación de la frecuencia de incidencia: Muy común (mayor o igual a 1/10), común (mayor o igual a 1/100 y menor que 1/10), no común (mayor o igual a 1/1.000 y menor que 1/100), rara (mayor o igual a 1/10.000 y menor que 1/1.000) y muy rara (menor que 1/10.000). Se ha notificado que los efectos adversos que se presentan a continuación se encuentran asociados con Tykerb®:
Ver Tabla
Se ha notificado que los efectos adversos adicionales que se presentan a continuación se encuentran asociados con Tykerb®, administrado en combinación con capecitabina, con una diferencia en la frecuencia de incidencia de más del 5%, en comparación con la frecuencia de incidencia observada con la capecitabina sola. Estas informaciones están basadas en la exposición a esta combinación en 198 pacientes.
Ver Tabla
Además, se notificó que los efectos adversos que se presentan a continuación se encuentran asociados con Tykerb®, administrado en combinación con capecitabina, pero se observaron a una frecuencia de incidencia similar a la del grupo que recibió monoterapia con capecitabina.
Ver Tabla
Se ha reportado que las siguientes reacciones adversas adicionales están asociadas con la administración de Tykerb® en combinación con letrozol, con una diferencia de más de 5% en la incidencia, en comparación con la monoterapia con letrozol. Estos datos se basan en la exposición a esta combinación en 654 pacientes.
Ver Tabla
|
|
Contraindicaciones:
Tykerb® está contraindicado en pacientes con hipersensibilidad a cualquiera de los ingredientes (ver Efectos colaterales).
|
|
Advertencias:
Tykerb® ha sido asociado con reportes de disminución en la fracción de eyección del ventrículo izquierdo [FEVI] (ver Efectos colaterales). Se deberá proceder con precaución cuando se requiera administrar Tykerb® a pacientes con condiciones que pudieran alterar la función del ventrículo izquierdo. Antes de iniciar el tratamiento con Tykerb®, se deberá evaluar la FEVI en todas las pacientes para garantizar que tengan una FEVI basal que se encuentre dentro de los límites institucionales normales. Durante todo el tratamiento con Tykerb®, se deberá seguir evaluando la FEVI para garantizar que no disminuya a un nivel inaceptable (ver Posología - demora y reducción en la dosificación - eventos cardíacos y Estudios Clínicos). La terapia con Tykerb® ha sido asociada con reportes de enfermedad pulmonar intersticial/neumonitis (ver Efectos colaterales). Se deberá vigilar y monitorizar a las pacientes en busca de síntomas pulmonares que sean sugestivos de enfermedad pulmonar intersticial/neumonitis (ver Posología). Se ha observado hepatotoxicidad (ALT o AST >3 veces el límite superior del normal y bilirrubina total >1.5 veces el límite superior del normal) en estudios clínicos (<1% de los pacientes) y en la experiencia posterior a la comercialización. La hepatotoxicidad podría ser severa y se han notificado muertes, aunque su relación con Tykerb® es incierta. La hepatotoxicidad podría presentarse en cuestión de días, o varios meses después del inicio del tratamiento. Se deben vigilar las pruebas de función hepática (transaminasas, bilirrubina y fosfatasa alcalina) antes de iniciar el tratamiento, cada 4 a 6 semanas durante el tratamiento, y cuando sea clínicamente indicado. Si se presentan cambios severos en la función hepática, se deberá suspender la terapia con Tykerb® y no volver a tratar a estos pacientes con este fármaco (ver Efectos colaterales). Si se va a administrar Tykerb® a pacientes con insuficiencia hepática severa preexistente, se recomienda reducir la dosis. En pacientes que desarrollen hepatotoxicidad severa mientras se encuentran bajo terapia, se deberá suspender la terapia con Tykerb® y no volver a tratar a estos pacientes con este fármaco (ver Posología y Farmacocinética - Poblaciones de Pacientes Especiales). Se han notificado casos de diarrea, incluyendo diarrea severa, al administrar el tratamiento con Tykerb® (ver Efectos colaterales). Es importante que se instituya un tratamiento proactivo de la diarrea con antidiarreicos. Los casos de diarrea severa podrían requerir la administración de líquidos y electrolitos orales o I.V., así como la interrupción o suspensión del tratamiento con Tykerb® (ver Posología -Demora y reducción en la dosificación - otras toxicidades). El tratamiento concomitante con inhibidores o inductores de la isoenzima CYP3A4 debe hacerse con precaución debido al riesgo de aumento o disminución, respectivamente, en la exposición a lapatinib (ver Interacciones). Debe evitarse la coadministración de Tykerb® con medicamentos con rangos terapéuticos estrechos que sean substratos de las isoenzimas CYP3A4 y CYP2C8 (véase Interacciones).
|
|
Precauciones:
Embarazo y lactancia: Fertilidad: No hay información relevante. Embarazo: No se han realizado estudios adecuados y bien controlados de Tykerb® en mujeres embarazadas. Se desconoce el efecto que ejerce Tykerb® en el embarazo humano. Tykerb® deberá administrarse durante el embarazo sólo si los beneficios esperados justifican el riesgo potencial para el feto. Se debe aconsejar a las mujeres con capacidad de procrear que utilicen métodos anticonceptivos adecuados y eviten quedar embarazadas mientras se encuentren recibiendo el tratamiento con Tykerb®. Cuando se estudió en ratas y conejas preńadas, Tykerb® no fue teratogénico pero ocasionó anormalidades menores al administrarse a dosis tóxicas para las madres (ver Información no clínica). Lactancia: Se desconoce si Tykerb® se excreta en la leche materna humana. Como muchos fármacos se excretan en la leche materna humana, y debido al riesgo potencial de efectos adversos ocasionados por Tykerb® en lactantes cuyas madres reciben este medicamento, se recomienda suspender el amamantamiento en aquellas mujeres que se encuentren recibiendo algún tratamiento con Tykerb®. Efectos sobre la capacidad de conducir y operar maquinaria: No se han realizado estudios para investigar el efecto que ejerce Tykerb® en la capacidad de conducir vehículos u operar maquinaria. Con base en el perfil farmacológico de Tykerb®, no es posible predecir efecto perjudicial alguno en dichas actividades. Al momento de considerar la capacidad del paciente de desempeńar tareas que requieren discernimiento y habilidades psicomotoras o cognoscitivas, se deberá tener presente su estado clínico y el perfil de efectos adversos de Tykerb®.
|
|
Interacciones Medicamentosas:
El lapatinib se metaboliza principalmente a través de la isoenzima CYP3A (véase Farmacocinética). Por tanto, los inhibidores o inductores de estas enzimas son capaces de alterar el perfil farmacocinético del lapatinib. La coadministración de Tykerb® con inhibidores conocidos de la isoenzima CYP3A4 (por ej.: ketoconazol, itraconazol, jugo de pomelo) debe hacerse con precaución, debiéndose monitorear cuidadosamente la respuesta clínica y los eventos adversos (ver Advertencias y Precauciones). Si concomitantemente los pacientes deben recibir un potente inhibidor de la CYP3A4, basados en estudios farmacocinéticos, debe tomarse en cuenta que se estima que una reducción en la dosis de Tykerb® a 500 mg/día podrá lograr los valores esperados para el Area Bajo la Curva (ABC) de lapatinib en condiciones en que los pacientes no hubieran recibido el potente inhibidor de la CYP3A4. Sin embargo, no hay datos clínicos con este ajuste de dosis en pacientes recibiendo inhibidores potentes de la CYP3A4. Si tal inhibidor potente se descontinúa, debe tomarse un período de lavado de aproximadamente 1 semana antes de aumentar la dosis de Tykerb® para alcanzar la recomendada. La coadministración de Tykerb® con inductores conocidos de CYP3A4 (por ej.: rifampina, carbamazepina o fenitoína) deberá realizarse con precaución, vigilando cuidadosamente la respuesta clínica y los eventos adversos (ver Advertencias y Precauciones). Si concomitantemente los pacientes deben recibir un potente inductor de la CYP3A4, basados en estudios farmacocinéticos, debe tomarse en cuenta que la dosis de Tykerb® debe aumentarse gradualmente entre 1250 mg/día y hasta 4500 mg/día, o de 1500 mg/día a 5500 mg/día, según la tolerabilidad. Se estima que esta dosis podrá lograr los valores esperados para el Área Bajo la Curva (ABC) de Tykerb® al rango esperado en condiciones en que los pacientes no hubieran recibido el inductor. Sin embargo, no hay datos clínicos con este ajuste de dosis en pacientes recibiendo potentes inductores de la CYP3A4. Si tal inductor potente se descontinúa, la dosis de Tykerb® debe reducirse durante aproximadamente 2 semanas para alcanzar la recomendada. In vitro, lapatinib inhibe la isoenzima CYP3A4 a concentraciones clínicamente relevantes. La coadministración de TYKERB con midazolam administrado vía oral produjo un incremento de aproximadamente 45% en el AUC del midazolam. No se observaron incrementos clínicamente significativos en el AUC cuando el midazolam se administró por vía intravenosa. Se deberá tener precaución al administrar Tykerb® de manera concurrente con medicamentos de rango terapéutico estrecho que sean sustratos de la isoenzima CYP3A4 (ver Farmacocinética).El lapatinib inhibe a la isoenzima CYP2C8 in vitro a concentraciones clínicamente relevantes. Se debe tener precaución al administrar Tykerb de manera concurrente con medicamentos con un estrecho intervalo terapéutico que sean sustratos de la CYP2C8 (véase Farmacocinética). La coadministración de Tykerb con paclitaxel intravenoso aumentó el nivel de exposición al paclitaxel en 23%, debido a la inhibición de la CYP2C8 y/o la P-glucoproteína (Pgp) a cargo del lapatinib. En estudios clínicos, se ha observado un incremento en la incidencia y severidad de casos de diarrea y neutropenia con esta combinación. Se recomienda tener precaución si se coadministra Tykerb con paclitaxel. La coadministración de Tykerb con docetaxel administrado vía intravenosa no afectó significativamente el AUC o la Cmax de alguna de las sustancias activas. Sin embargo, hubo un incremento en la incidencia de neutropenia inducida por docetaxel. La coadministración de Tykerb con irinotecán (cuando es administrado como parte del régimen de FOLFIRI) produjo un incremento de aproximadamente 40% en el AUC del SN 38, el metabolito activo del irinotecán. Se desconoce el mecanismo preciso de esta interacción. Se recomienda tener precaución si se coadministra Tykerb con irinotecán. Lapatinib es un sustrato para las proteínas transportadoras Pgp y BCRP. Los inhibidores e inductores de estas proteínas pueden alterar la disponibilidad y/o la distribución de lapatinib (ver Farmacocinética). Lapatinib inhibe la proteína transportadora Pgp in vitro a concentraciones clínicamente relevantes. La coadministración de Tykerb con digoxina administrada por vía oral produjo un incremento de aproximadamente 98% en el AUC de la digoxina. Se debe tener precaución al administrar Tykerb de manera concurrente con medicamentos con un estrecho intervalo terapéutico que sean sustratos de la Pgp. In vitro lapatinib inhibe a las proteínas transportadoras Pgp, BCRP y OATP1B1. La relevancia clínica de este efecto no se ha evaluado. No puede excluirse que lapatinib afectará la farmacocinética de los sustratos de Pgp (por ej.: Digoxina), BCRP (por ej.: topotecán) y OATP1B1 (por ej.: rosuvastatina) (ver Farmacocinética). La coadministración de Tykerb® con capecitabina, letrozol o trastuzumab no alteró significativamente el perfil farmacocinético de estos agentes (ni de los metabolitos de capecitabina) o lapatinib. La biodisponibilidad de lapatinib es afectada por los alimentos (ver Posología y Farmacocinética).
|
|
Sobredosificación:
No existe antídoto específico para la inhibición de la fosforilación de tirosina del ErbB1 (EGFR) y/o Erb B2 (HER2). La dosis oral máxima de Tykerb® que se ha administrado en estudios clínicos consiste en 1800 mg 1 vez al día. La ingestión de Tykerb® a una mayor frecuencia podría producir concentraciones séricas que excedan las observadas en los estudios clínicos. Por tanto, no se deberán reemplazar las dosis omitidas, sino reanudar la dosificación con la siguiente dosis diaria programada (ver Posología). Síntomas y signos: Casos asintomáticos y sintomáticos de sobredosis se han reportado en pacientes iniciando el tratamiento con Tykerb®. Los síntomas observados incluyen eventos conocidos asociados a Tykerb® (veáse Efectos colaterales) y en algunos casos dolor en el cuero cabelludo, taquicardia sinusal (con ECG normal) y/o inflamación de la mucosa. Tratamiento: Tykerb® exhibe un alto grado de fijación a proteínas plasmáticas y no experimenta una eliminación renal significativa. Por tanto, no se esperaría que la hemodiálisis constituyera un método eficaz para mejorar la eliminación de Tykerb®. El tratamiento ulterior deberá ser el indicado por el médico o el recomendado por el centro nacional de toxicología, donde esté disponible.
|
|
Incompatibilidades:
Sin incompatibilidades conocidas.
|
|
Conservación:
Precauciones especiales de almacenamiento: No almacenar a temperatura superior a 30ş C. Vida útil: la fecha de caducidad se indica en el empaque.
|
|
Presentaciones:
Envase conteniendo 70 comprimidos recubiertos.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}