 |
Composición:
Wellbutrin® XL 300 mg: Cada comprimido contiene: 300 mg de Clorhidrato de Anfebutamona (bupropión). Excipientes: Núcleo del comprimido: Alcohol Polivinílico; Behenato de Glicerilo. Recubrimiento del comprimido: Etilcelulosa 100; Macrogol 1450; Povidona; Dispersión de Copolímero de Acido Metacrílico Tipo C (Eudragit L30 D-55); Dióxido de Silicio; Citrato de Trietilo; Tinta Negra para Impresión. Presentación farmacéutica: comprimidos recubiertos de liberación extendida.
|
|
Acción Terapéutica:
Grupo farmacoterapéutico: Otros antidepresivos. Código ATC: N06 AX12.
|
|
Indicaciones:
Trastorno depresivo mayor: La formulación Wellbutrin® XL se indica en el tratamiento de episodios depresivos mayores. Después de haber obtenido una respuesta satisfactoria, la continuación de la terapia con Wellbutrin® XL resulta eficaz para prevenir una recaída. Trastorno afectivo estacional: Wellbutrin® XL está indicado en la prevención de los episodios de depresión mayor estacional en pacientes que tienen diagnóstico de trastorno afectivo estacional. El desorden afectivo estacional se caracteriza por episodios depresivos mayores recurrentes, que ocurren comúnmente durante los meses de otońo e invierno. Los episodios duran alrededor de 6 meses, iniciándose típicamente en otońo y remiten en la época de primavera. Aunque los pacientes con trastorno afectivo estacional pueden tener episodios depresivos durante otras estaciones del ańo, el diagnóstico de trastorno afectivo estacional requiere que el número de episodios estacionales substancialmente excedan el número de episodios no estacionales, a lo largo de la vida del paciente.
|
|
Propiedades:
Farmacodinamia: El bupropión es un inhibidor selectivo de la recaptación neuronal de catecolaminas (noradrenalina y dopamina), el cual exhibe un efecto mínimo en la recaptación de indolaminas (serotonina) y no inhibe la monoaminoxidasa. Como se desconoce el mecanismo de acción del bupropión, al igual que el de otros antidepresivos, se supone que su acción es mediada por mecanismos noradrenérgicos y/o dopaminérgicos. En un estudio realizado en voluntarios sanos, no se observaron efectos clínicamente significativos en el intervalo QTcF al administrar comprimidos de bupropión de liberación extendida (450 mg/día), en comparación con la administración de placebo, después de 14 días de administración hasta alcanzar el estado estacionario. Farmacocinética: Absorción: Después de la administración oral de Wellbutrin® XL a voluntarios sanos, se lograron concentraciones plasmáticas máximas de anfebutamona (bupropión) después de 5 horas. La absorción de los comprimidos de anfebutamona (bupropión) de liberación extendida no se ve afectada significativamente cuando se ingieren con alimentos. La anfebutamona (bupropión) y sus metabolitos muestran una cinética lineal después de la administración crónica de 150 a 300 mg diarios. Distribución: La anfebutamona (bupropión) se distribuye ampliamente, con un volumen aparente de distribución de aproximadamente 2000 L. El bupropión y el hidroxibupropión se fijan moderadamente a las proteínas plasmáticas (84% y 77%, respectivamente). El grado de fijación proteica del metabolito treohidrobupropión es de aproximadamente la mitad del observado con el bupropión. Metabolismo: La anfebutamona (bupropión) se metaboliza ampliamente en los humanos. En el plasma, se han identificado 3 metabolitos farmacológicamente activos: hidroxibupropión y los isómeros de amino-alcohol, treohidrobupropión y eritrohidrobupropión. Es posible que éstos tengan alguna importancia clínica, ya que sus concentraciones plasmáticas son tan altas, o más altas, que las del bupropión. Las concentraciones plasmáticas máximas de hidroxibupropión y treohidrobupropión se alcanzan aproximadamente 6 horas después de administrar una dosis única de Wellbutrin® XL. No se pueden medir las concentraciones de eritrohidrobupropión en el plasma después de administrar una dosis única de Wellbutrin® XL. A su vez, los metabolitos activos se metabolizan a metabolitos inactivos, los cuales se excretan en la orina. Los estudios in vitro indican que el bupropión se metaboliza a su metabolito activo principal, el hidroxibupropión, a través de la isoenzima CYP2B6, principalmente, mientras que los citocromos P450 no están implicados en la formación de treohidrobupropión (véase Interacciones). Tanto el bupropión como el hidroxibupropión son inhibidores competitivos, relativamente débiles, de la isoenzima CYP2D6 con Ki valores de 21 y 13.3 µM, respectivamente. En voluntarios humanos identificados como amplios metabolizadores de la isoenzima CYP2D6, la administración concomitante de bupropión y desipramina ocasionó aumentos de 2 y 5 veces en la Cmáx y el ABC de desipramina, respectivamente. Este efecto estuvo presente durante cuando menos 7 días después de administrar la última dosis de bupropión. Puesto que el bupropión no se metaboliza por la vía de la CYP2D6, no se pronostica que la desipramina afecte la farmacocinética del bupropión. Se recomienda tener precaución al administrar Wellbutrin® XL con sustratos para la vía CYP2D6 (véase Interacciones). Se ha demostrado que el bupropión induce su propio metabolismo en animales, después de llevar a cabo una administración casi crónica. En los humanos, no hay indicios de inducción enzimática de bupropión o hidroxibupropión en pacientes o voluntarios que recibieron las dosis recomendadas de bupropión durante 10 a 45 días. En un estudio realizado en voluntarios sanos, el ritonavir administrado a una dosis de 100 mg 2 veces al día redujo el AUC y la Cmax del bupropión en 22% y 21%, respectivamente. El AUC y la Cmax de los metabolitos del bupropión experimentaron un decremento de 0 a 44%. En un segundo estudio realizado en voluntarios sanos, el ritonavir administrado a una dosis de 600 mg 2 veces al día redujo el AUC y la Cmax del bupropión en 66% y 62%, respectivamente. El AUC y la Cmax de los metabolitos del bupropión experimentaron un decremento de 42 a 78%. En otro estudio realizado en voluntarios sanos, la administración de Kaletra® (400 mg de lopinavir/100 mg de ritonavir 2 veces al día) redujo el AUC y la Cmáx del bupropión en 57%. El AUC y la Cmax del hidroxibupropión experimentaron un decremento de 50% y 31%, respectivamente. Eliminación: Después de la administración oral de 200 mg de 14C-bupropión en humanos, se recuperó el 87% y 10% de la dosis radioactiva en la orina y heces, respectivamente. La fracción de la dosis de bupropión que se excretó en forma inalterada sólo representó un 0.5%, lo cual es un hallazgo que coincide con el amplio metabolismo del bupropión. Menos del 10% de esta dosis de 14C fue cuantificada en la orina como metabolitos activos. La depuración media aparente, posterior a la administración oral de bupropión, es de aproximadamente 200 l/hr y el promedio de la vida media de eliminación de bupropión es de aproximadamente 20 horas. La vida media de eliminación del hidroxibupropión es de aproximadamente 20 horas, y su área bajo la curva de tiempo frente a concentración plasmática de fármaco (ABC), en estado estacionario, es de aproximadamente 17 veces la del bupropión. Las vidas medias de eliminación del treohidrobupropión y el eritrohidrobupropión son más prolongadas (37 y 33 horas, respectivamente) y los valores del ABC en estado estacionario son 8 y 1.6 veces mayores que los del bupropión, respectivamente. El estado estacionario para el bupropión y sus metabolitos se alcanza en 8 días. Pacientes con insuficiencia renal: La eliminación de bupropión y sus principales metabolitos puede verse afectada por una reducción en la función renal (consulte Advertencias y precauciones especiales para su uso). En sujetos con insuficiencia renal de grado moderado a severo o en etapa terminal, se observó un incremento en el nivel de exposición al bupropión y/o sus metabolitos. Pacientes con insuficiencia hepática: La farmacocinética del bupropión y sus metabolitos activos no presentó diferencias estadísticamente significativas en los pacientes con cirrosis de grado leve a moderado, en comparación con voluntarios sanos, aunque se observó una mayor variabilidad entre los pacientes individuales. En los pacientes con cirrosis hepática grave, la Cmáx y el ABC del bupropión aumentaron sustancialmente (diferencia media aproximada de 70% y 3 veces, respectivamente) y tuvieron una mayor variabilidad, al compararse con los valores de los voluntarios sanos; la vida media promedio también fue más prolongada (aproximadamente 40%). Para los metabolitos, la Cmáx media fue más baja (aproximadamente 30 a 70 %), el ABC medio tendió a ser más alto (aproximadamente 30 a 50 %), el Tmáx mediano fue tardío (aproximadamente 20 hrs), y las vidas medias promedio fueron más prolongadas (aproximadamente, 2 a 4 veces) que en voluntarios sanos (véase Advertencias y Precauciones). Pacientes de edad avanzada: Los estudios farmacocinéticos realizados en pacientes de edad avanzada han mostrado resultados variables. En un estudio realizado con dosis únicas, se demostró que la farmacocinética del bupropión y sus metabolitos, observada en pacientes de edad avanzada, no difiere de aquella que exhiben los adultos más jóvenes. Otro estudio farmacocinético, realizado con dosis tanto únicas como múltiples, sugirió que los pacientes de edad avanzada podrían experimentar una mayor acumulación de bupropión y sus metabolitos. La experiencia clínica existente no ha identificado alguna diferencia en la tolerabilidad exhibida entre ancianos y pacientes más jóvenes, pero no se puede descartar que los pacientes mayores experimenten una mayor sensibilidad. Estudios clínicos: La eficacia de anfebutamona en el tratamiento del desorden depresivo mayor fue establecida usando formulaciones de liberación convencional en un ensayo clínico controlado con placebo de 4 semanas de duración, realizado en pacientes hospitalizados, y en otro ensayo clínico placebo controlado de 6 semanas de duración en pacientes ambulatorios. En el primer estudio, los pacientes recibieron 300 a 600 mg/día de una formulación de liberación convencional de anfebutamona clorhidrato, administrados en 3 dosis diarias. Este estudio demostró la efectividad de anfebutamona al usar la escala de Hamilton para depresión (HDRS) y la escala de impresión clínica global (CGI) de severidad. El segundo estudio fue realizado con 2 dosis de una formulación de liberación convencional de anfebutamona (350 mg y 450 mg) y placebo. Se demostró la eficacia de anfebutamona, al usar las escalas HDRS y CGI de severidad, pero no así para el ítem 1 de la escala HDRS, que corresponde a conducta depresiva. Un tercer estudio realizado en pacientes ambulatorios, demostró la eficacia de una dosis de 300 mg de una formulación de anfebutamona de liberación convencional. La efectividad se demostró en base a la escalas HDRS, el ítem 1 de HDRS, la escala de Montgomery-Asbertg para depresión, CGI para severidad, CGI para mejoramiento. En un estudio a largo plazo, realizado en pacientes ambulatorios que cumplían el criterio DSM-IV para desorden depresivo mayor de tipo recurrente y que durante 8 semanas estuvieron sometidos a un ensayo abierto, recibiendo 150 mg 2 veces al día de anfebutamona (formulación SR de liberación sostenida), fueron randomizados en 2 grupos, uno de los cuales continuó recibiendo la misma dosis de anfebutamona, y el otro grupo fue asignado a placebo. Se observó las recaídas hasta por un período de 44 semanas. Los pacientes que recibieron la formulación SR presentaron menores índices de recaídas, respecto placebo. Aunque no se han realizado estudios independientes para avalar la efectividad antidepresiva de Wellbutrin® XL, se han demostrado bajo condiciones de estado estacionario, la bioequivalencia de Wellbutrin® XL con formulaciones de liberación convencional y con formulaciones de liberación sostenida (SR), es decir Wellbutrin® XL 300 mg 1 vez al día es bioequivalente a 100 mg 3 veces al día de la formulación de liberación convencional, y a 150 mg 2 veces al día de la formulación SR, respecto a la concentración plasmática máxima y grado de absorción. Trastorno afectivo estacional: La eficacia de Wellbutrin® XL para la prevención de episodios de depresión mayor estacional asociados con trastorno afectivo estacional fue establecida en 3 estudios doble ciego, placebo controlado, en adultos ambulatorios con una historia de trastorno depresivo mayor durante la época de otońo-invierno (como se encuentra definido por criterios de la DSM-IV). El tratamiento fue iniciado en el otońo del hemisferio norte, antes del inicio de los síntomas (septiembre - noviembre) y fue discontinuado después de 2 semanas tras una disminución gradual, luego de la primera semana del inicio de la primavera (4Ş semana de marzo), resultando una duración de tratamiento de aproximadamente 4 a 6 meses para la mayoría de los pacientes. Al inicio del estudio los pacientes fueron randomizados para recibir placebo o Wellbutrin® XL 150 mg 1 vez al día, por 1 semana, seguidos de una titilación a 300 mg 1 vez al día. Los pacientes quienes fueron considerados por el investigador, como poco probables o incapaces de tolerar una dosis de 300 mg 1 vez al día, se les permitió mantener o reducir la dosis a 150 mg 1 vez al día. El promedio de las dosis de Wellbutrin® XL en los 3 estudios fue desde 257 mg a 280 mg/día. En estos 3 ensayos, el porcentaje de pacientes que estuvieron sin depresión al final del tratamiento, fue significativamente más alto para Wellbutrin® XL versus placebo: 81.4% vs 69.7%; 87.2% vs 78.8% y 84.0% vs 69.0% para los estudios 1, 2 y 3 respectivamente, con una tasa libre depresión en los 3 estudios combinados de 84.3% vs 72.0%. Datos preclínicos de seguridad: Carcinogénesis/mutagénesis: Los estudios de oncogenicidad realizados en ratas y ratones confirman la ausencia de carcinogenicidad en estas especies. Toxicología reproductiva: Fertilidad: No hubo evidencia de disminución de la fertilidad en ratas a dosis de hasta aproximadamente 7 veces la dosis máxima recomendada para humanos (DMRH) sobre una base de mg/m2. Embarazo: No hubo evidencia de teratogenicidad en ratas o conejos a dosis de hasta aproximadamente 11 y 7 veces la DMRH, respectivamente, basada en una base de mg/m2 (la exposición a la dosis alta en uno de los estudios con ratas, 300 mg/kg/día, fue 1.7-veces que la humana basándose en los valores AUC en estado de equilibrio). En conejos, se observó un ligero aumento en las variaciones esqueléticas (aumento de la incidencia de variación anatómica común de una costilla torácica accesoria y retraso de la osificación de falanges) a dosis aproximadamente iguales a la dosis máxima humana y mayores, y disminuyó el peso fetal a dosis tóxicas para la madre. A exposiciones hasta de aproximadamente 7 veces la DMRH sobre la base de mg/m2 no se observaron efectos adversos en los productos de ratas administradas con bupropión antes del acoplamiento y durante el embarazo y la lactancia. Toxicología animal y/o farmacología: En estudios realizados en animales, se observaron cambios hepáticos, pero éstos reflejan la acción de un inductor de enzimas hepáticas. A las dosis clínicas administradas en humanos, no existen indicios de inducción enzimática alguna, lo cual sugiere que los hallazgos hepáticos en animales de laboratorio sólo tienen una importancia limitada en la evaluación y valoración de los riesgos asociados con el bupropión.
|
|
Posología:
Los comprimidos de Wellbutrin® XL deben deglutirse enteros, sin partirse ni masticarse, ya que esto podría incrementar el riesgo de que el paciente desarrolle efectos adversos, incluyendo convulsiones. Los comprimidos de Wellbutrin® XL pueden tomarse con o sin alimentos. Trastorno depresivo mayor: Uso en adultos: La dosis única máxima de Wellbutrin® XL consiste en 450 mg. El insomnio es un efecto adverso muy común, que por lo general es transitorio. El insomnio puede reducirse si se evita la administración a la hora de acostarse (siempre y cuando haya un intervalo de cuando menos 24 horas entre las dosis) o, si está indicado clínicamente, reduciendo la dosificación. Tratamiento inicial: La dosis habitual es de 300 mg 1 vez al día en la mańana. La dosis inicial de los comprimidos de anfebutamona (bupropión) de liberación extendida consiste en 150 mg, administrados por la mańana como una dosis diaria única. Si la dosis de 150 mg/día se tolera, un incremento a 300 mg/día, administrados 1 vez al día puede ser realizada a partir del cuarto día de dosificación. Se debe conservar un intervalo de al menos 24 horas entre dosis sucesivas. Tal como ocurre con otros antidepresivos, es posible que el efecto máximo del Wellbutrin® XL no sea evidente sino hasta después de varias semanas de tratamiento. Aumento de la dosis por sobre los 300 mg/día: Un aumento en la dosificación de bupropión hasta 450 mg/día, dado como dosis única, puede ser beneficioso en pacientes que no han mostrado una mejoría clínica después de varias semanas de tratamiento con Wellbutrin® XL de 300 mg/día. Transferencia de pacientes bajo tratamiento con Wellbutrin® SR comprimidos: Al transferir pacientes bajo terapia con Wellbutrin® SR comprimidos a un tratamiento con Wellbutrin® XL comprimidos, en lo posible se deberá administrar la misma dosis total diaria. Los pacientes que en la actualidad reciben tratamiento con Wellbutrin® SR comprimidos, a razón de 300 mg/día (por ejemplo, 150 mg administrados 2 veces al día), pueden ser transferidos a un régimen de tratamiento de 300 mg de Wellbutrin® XL administrados 1 vez al día. Terapia de mantenimiento: Es de consenso general que los pacientes que presentan episodios de depresión aguda requieren 6 meses, o más, de tratamiento con fármacos antidepresivos. No se sabe si la dosis necesaria de Wellbutrin®XL para el tratamiento de mantenimiento sea idéntica a la dosis requerida para alcanzar una respuesta inicial. En consecuencia, los pacientes deberán ser reevaluados para determinar la necesidad de un tratamiento de mantención y la dosis para tal efecto. Trastorno afectivo estacional: Para la prevención de episodios depresivos mayor estacional asociados con el trastorno afectivo estacional, la dosificación de Wellbutrin® XL debe ser iniciada generalmente en otońo, antes del inicio de los síntomas depresivos. El tratamiento debe ser continuado durante la época de invierno y deberá ser gradualmente discontinuado a inicios de la primavera. Los tiempos de iniciación y duración del tratamiento deben ser individualizados según los patrones históricos de episodios de depresión mayor estacional del paciente. Los pacientes cuyos episodios depresivos estacionales son infrecuentes o no están asociados con dańos significativos, no deben ser tratados profilácticamente. La dosificación con Wellbutrin® XL debe comenzar con 150 mg al día, administrado en una dosis simple en la mańana. Si los 150 mg diarios en la mańana son tolerados adecuadamente, la dosis debe ser aumentada a 300 mg al día después de 1 semana. Si los 300 mg no fueran tolerados adecuadamente, podrían reducirse a 150 mg al día. Para el adulto, la dosis habitual de Wellbutrin® XL es de 300 mg al día, administrado 1 vez al día en la mańana. Para los pacientes que están tomando 300 mg al día durante la época de otońo e invierno, la dosis debe ser gradualmente disminuida a 150 mg al día, por 2 semanas, antes de la discontinuación. Las dosis de Wellbutrin® XL por sobre los 300 mg al día, no han sido estudiadas para la prevención de episodios depresivos mayor estacional. Uso en nińos y adolescentes: El uso de Wellbutrin® XL no se indica en nińos o adolescentes menores de 18 ańos de edad (véase Advertencias y Precauciones). Aún no se establecen la seguridad y la eficacia de la administración de Wellbutrin® XL a pacientes menores de 18 ańos de edad. Uso en pacientes de edad avanzada: No es posible descartar la posibilidad de que algunos individuos de edad avanzada experimenten una mayor sensibilidad al bupropión, por lo cual se podría requerir una reducción en la frecuencia y/o la dosis (véase Advertencias y Precauciones). Uso en pacientes con insuficiencia hepática: Wellbutrin® XL debe emplearse con precaución en los pacientes con insuficiencia hepática. Debido a que los pacientes con cirrosis hepática de leve a moderada experimentan una mayor variabilidad en la farmacocinética, se debe considerar una reducción en la dosis y/o frecuencia de la administración (véase Advertencias y Precauciones). Wellbutrin® XL debe emplearse con extrema precaución en los pacientes con cirrosis hepática grave. En estos pacientes, la dosis no debe exceder 150 mg administrados en días alternos (véase Advertencias y Precauciones). Uso en pacientes con insuficiencia renal: En estos pacientes, la anfebutamona (bupropión) y sus metabolitos activos podrían experimentar una mayor acumulación en dichos pacientes (véase Advertencias y Precauciones), por tanto se debe administrar con precaución a una dosis y/o frecuencia reducidas.
|
|
Efectos Colaterales:
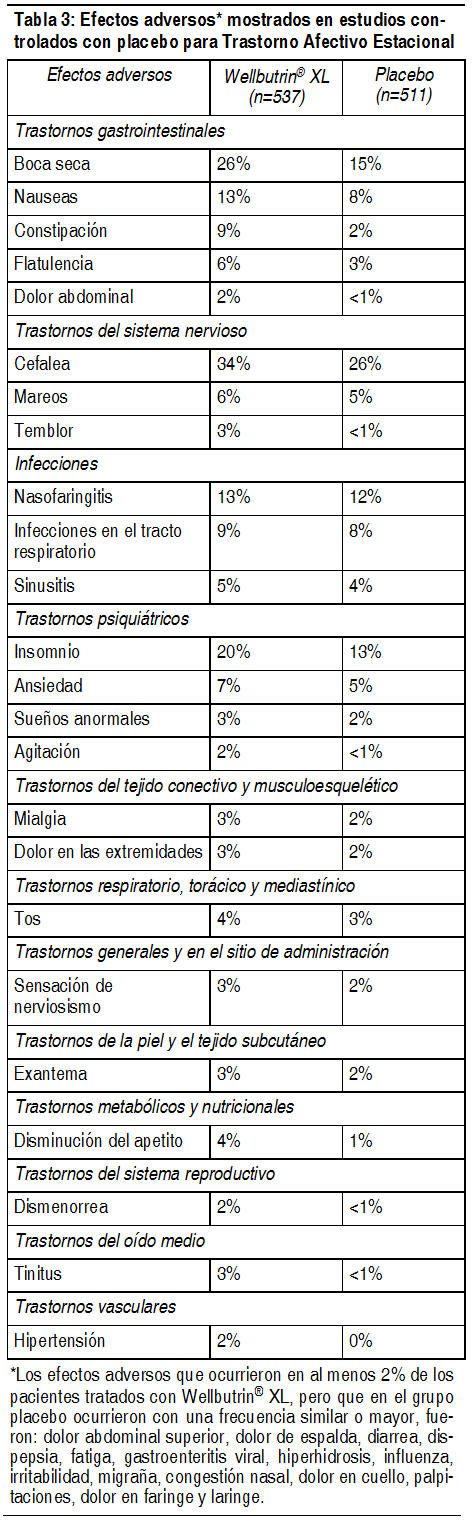
La siguiente lista proporciona información sobre los efectos adversos, clasificados por sistema corporal, que se identificaron a partir de la experiencia clínica. Generales: Fiebre, dolor torácico, astenia. Cardiovasculares: Taquicardia, palpitaciones, vasodilatación, hipotensión ortostática, elevaciones en la presión arterial (en algunos casos severas), rubefacción, síncope. SNC: Convulsiones (véase Advertencias y Precauciones), insomnio, temblores, distonía, ataxia, parkinsonismo, espasmos, falta de coordinación, trastornos en la concentración, cefalea, mareos, depresión, confusión, delirios, ideación paranoide, alucinaciones, agitación, inquietud, ansiedad, irritabilidad, hostilidad, agresividad, despersonalización, sueńos anormales, alteración de la memoria, parestesia. Endocrinos y metabólicos: Anorexia, pérdida de peso, trastornos de la glucemia. Gastrointestinales: Boca seca, trastornos gastrointestinales incluyendo náuseas y vómito, dolor abdominal, estreńimiento. Genitourinarios: Polaquiuria y/o retención urinaria. Hepatobiliares: Elevación de enzimas hepáticas, ictericia, hepatitis. Cutáneos/hipersensibilidad: Exantema, prurito, sudación. Reacciones de hipersensibilidad cuya severidad varía de urticaria a angioedema, disnea/broncoespasmo y, en raras ocasiones, shock anafiláctico. También se han producido comunicaciones de artralgia, mialgia y fiebre, en asociación con exantema y otros síntomas que sugieren una reacción de hipersensibilidad tardía. Es posible que estos síntomas asemejen la enfermedad del suero. En raras ocasiones, también se han comunicado casos de eritema multiforme y síndrome de Stevens-Johnson. Sentidos especiales: Acúfenos, trastornos visuales, trastornos del gusto. Eventos adversos asociados al trastorno afectivo estacional: Efectos adversos que llevaron a la discontinuación del tratamiento: En estudios clínicos, controlados con placebo, el 9% de pacientes tratados con Wellbutrin® XL y el 5% de los pacientes tratados con placebo, discontinuaron el tratamiento debido a los efectos adversos presentados. Los efectos adversos que condujeron a la interrupción del tratamiento en al menos 1% de los pacientes tratados con Wellbutrin® XL, correspondiente a una tasa numéricamente mayor que la del placebo, fueron insomnio (2 % contra < el 1%) y cefalea (1% contra < el 1%). Efectos colaterales que ocurren en una incidencia del 2% o más en pacientes tratados con Wellbutrin® XL: La tabla 3 enumera los efectos adversos inesperados, encontrados en 3 estudios controlados con placebo de pacientes tratados con Wellbutrin® XL durante 6 meses o más. Se incluyeron los efectos ocurridos en una incidencia del 2 % o más y que fueron más frecuentes que en el grupo de placebo. Los efectos colaterales fueron clasificados basándose en el MedDRA.
Ver Tabla
|
|
Contraindicaciones:
Wellbutrin® XL está contraindicado en aquellos pacientes con hipersensibilidad a la anfebutamona (bupropión), o a cualquiera de los componentes de la preparación. Wellbutrin® XL está contraindicado en pacientes que presenten crisis convulsivas. Wellbutrin® XL está contraindicado en los pacientes sometidos a una suspensión abrupta de alcohol o sedantes. Wellbutrin® XL en comprimidos contiene anfebutamona (bupropión), por lo cual no debe administrarse a pacientes que actualmente se encuentren bajo tratamiento con cualquier otra preparación que contenga la anfebutamona (bupropión), ya que la incidencia de crisis convulsivas depende de la dosis. Wellbutrin® XL se contraindica en los pacientes que presentan un diagnóstico actual o previo de bulimia o anorexia nerviosa, ya que en esta población de pacientes se observó una mayor incidencia de crisis convulsivas cuando se administró una formulación de anfebutamona (bupropión) de liberación inmediata. Se contraindica el uso concomitante de Wellbutrin® XL con inhibidores de la monoaminoxidasa (IMAOs). Deberán transcurrir por lo menos 14 días entre la suspensión de la terapia con IMAOs y el inicio del tratamiento con Wellbutrin® XL comprimidos.
|
|
Advertencias:
De acuerdo con la indicación del Wellbutrin® SR como antidepresivo, es importante considerar las siguientes advertencias: En un análisis de 24 estudios clínicos a corto plazo (4 meses), que involucraron a 4400 pacientes nińos con depresión mayor, desorden obsesivo compulsivo u otras alteraciones psiquiátricas, placebo controlados, quienes fueron tratados con antidepresivos inhibidores selectivos de la recaptación de serotonina y antidepresivos de otra clase, se observó un incremento del doble en riesgo de suicidio del grupo que recibió el antidepresivo versus el grupo que recibió placebo (4% versus 2%). Por lo tanto, se recomienda que antes de iniciar la terapia con un antidepresivo se deben investigar cuidadosamente los antecedentes psiquiátricos del paciente, incluyendo historia familiar y personal de suicidios y desorden bipolar. Convulsiones: No se debe exceder la dosis recomendada de Wellbutrin® XL, ya que el riesgo de sufrir convulsiones se asocia con la dosificación de anfebutamona (bupropión). En los estudios clínicos, la incidencia general de los casos de convulsiones asociados con Wellbutrin® SR, con dosis de hasta 300 mg/día, fue de aproximadamente 0.1%. Se debe suspender la terapia con Wellbutrin® XL, y no volverse a iniciar, en aquellos pacientes que experimenten alguna convulsión durante el tratamiento. El riesgo de que se presenten convulsiones al usar la anfebutamona (bupropión) parece asociarse fuertemente con la presencia de factores de riesgo predisponentes. Por tanto, Wellbutrin® XL debe administrarse con extrema precaución en pacientes con uno o más factores predisponentes para un bajo umbral convulsivo. Estos incluyen: Antecedente de traumatismo encéfalo-craneano. Tumores en el sistema nervioso central (SNC). Antecedentes de crisis convulsivas. Administración concomitante de otros medicamentos que se sabe disminuyen el umbral de convulsiones (por ejemplo: antipsicóticos, antidepresivos, teofilina, esteroides sistémicos). Cirrosis hepática severa. Además, se debe tener precaución en aquellas circunstancias clínicas asociadas con un aumento en el riesgo de convulsiones. Estas incluyen: uso excesivo de alcohol o sedantes (véase Contraindicaciones), diabetes tratada con hipoglucemiantes o insulina y el uso de estimulantes o productos anoréxicos. Wellbutrin® XL debe ser discontinuado y no recomendado en pacientes quienes experimenten convulsiones mientras están en tratamiento. Para disminuir el riesgo de convulsiones asociadas al uso de Wellbutrin® XL, se recomienda no sobrepasar la dosis diaria de 450 mg y los incrementos de dosis deben ser graduales. Reacciones de hipersensibilidad: La terapia con Wellbutrin® XL debe suspenderse inmediatamente si los pacientes experimentan reacciones de hipersensibilidad durante el tratamiento (véase Efectos colaterales). Los médicos deben estar conscientes de que los síntomas pueden persistir aun después de suspender la terapia con anfebutamona (bupropión). Además, se debe proporcionar un tratamiento médico, según sea el caso. Insuficiencia hepática: La anfebutamona (bupropión) se metaboliza ampliamente en el hígado a metabolitos activos, los cuales a su vez se metabolizan posteriormente. En comparación con voluntarios sanos, los pacientes con cirrosis hepática leve a moderada no mostraron diferencias estadísticamente significativas en la farmacocinética de la anfebutamona (bupropión), pero hubo una mayor variabilidad en las concentraciones plasmáticas de bupropión entre los pacientes individuales. Por tanto, Wellbutrin® XL debe emplearse con precaución en los pacientes con insuficiencia hepática y, asimismo, deberá considerarse una reducción en la dosis y/o frecuencia de administración en los pacientes con cirrosis hepática de leve a moderada (véase Posología). Wellbutrin® XL debe emplearse con extrema precaución en los pacientes con cirrosis hepática grave. Es necesario reducir la frecuencia de la administración en estos pacientes, ya que experimentan un aumento sustancial en las concentraciones máximas de bupropión, por lo que es probable que haya lugar a una mayor acumulación en estos pacientes (véase Posología). Se debe vigilar estrechamente a todos los pacientes con insuficiencia hepática, con el fin de determinar posibles efectos adversos (por ejemplo, insomnio, boca seca, convulsiones), los cuales podrían indicar concentraciones elevadas de fármaco o sus metabolitos. A altas dosis, se ha demostrado un potencial hepatotóxico en modelos animales. Insuficiencia renal: La anfebutamona (bupropión) se metaboliza ampliamente en el hígado a metabolitos activos, los cuales a su vez se metabolizan posteriormente y se excretan por la vía renal. Por tanto, el tratamiento de los pacientes con insuficiencia renal debe iniciarse a una dosis y/o frecuencia reducidas, ya que el bupropión y sus metabolitos podrían experimentar un mayor grado de acumulación en estos pacientes (ver Farmacocinética). Se debe vigilar estrechamente a los pacientes con el fin de determinar posibles efectos adversos (por ejemplo, insomnio, boca seca, convulsiones), los cuales podrían indicar concentraciones elevadas de fármaco o metabolitos. Pacientes de edad avanzada: La experiencia clínica existente con anfebutamona (bupropión) no ha identificado diferencia alguna entre la tolerabilidad por los pacientes de edad avanzada y los demás pacientes adultos. Sin embargo, no es posible descartar que algunos individuos de edad avanzada tengan una mayor sensibilidad al bupropión, por lo que es posible que se requiera reducir la frecuencia y/o la dosis de administración (véase Farmacocinética). Nińos y adolescentes <18 ańos de edad: El tratamiento con antidepresivos se asocia con un aumento en el comportamiento y pensamientos suicidas en nińos y adolescentes con trastorno depresivo mayor, así como otros trastornos psiquiátricos. La seguridad y la eficacia del uso de anfebutamona clorhidrato no han sido establecidas en la población pediátrica. Agravamiento clínico y riesgo de suicidio en adultos con trastornos psiquiátricos: Los pacientes deprimidos podrían experimentar un agravamiento de sus síntomas depresivos, o ideación y comportamiento suicida (tendencias suicidas), o ambas cosas, independientemente si se encuentran o no bajo tratamiento con medicamentos antidepresivos. El riesgo persiste hasta que se presenta una remisión significativa. Debido a que es posible que los pacientes no muestren alguna mejoría durante las primeras semanas o más de tratamiento, deben ser vigilados estrechamente con el fin de determinar si hay agravamiento clínico (incluyendo el desarrollo de nuevos síntomas) y tendencias suicidas, especialmente al inicio de un ciclo de tratamiento, o cuando se realicen cambios en la dosificación, ya sean aumentos o disminuciones. La experiencia clínica general existente con todas las terapias antidepresivas indica un posible aumento del riesgo de suicidio en los pacientes que se encuentran en las primeras etapas de recuperación. Los pacientes con antecedentes de comportamiento o pensamientos suicidas, adultos jóvenes y aquellos pacientes que presentan algún grado significativo de ideación suicida antes de comenzar el tratamiento, se encuentran en un mayor riesgo de experimentar pensamientos suicidas o intentos de suicidio, por lo cual deben ser vigilados cuidadosamente durante el tratamiento. Además, en un meta-análisis de estudios clínicos en los que se usaron antidepresivos, controlados con placebo, administrados a pacientes adultos con trastorno depresivo mayor y otros trastornos psiquiátricos, se demostró un incremento en el riesgo de desarrollar comportamientos y pensamientos suicidas asociados con el uso de antidepresivos, en comparación con el placebo, en pacientes menores de 25 ańos de edad. Se debe advertir a los pacientes (y a los que los cuidan) que es necesario establecer vigilancia para determinar cualquier agravamiento de sus trastornos (incluyendo el desarrollo de nuevos síntomas) y/o el surgimiento de ideación/comportamiento suicida o pensamientos de autoagresión, así como buscar asesoría médica en forma inmediata, en caso de que se presenten estos síntomas. Es preciso reconocer que la aparición de algunos síntomas neuropsiquiátricos podría relacionarse tanto como con el estado de la enfermedad subyacente, como con la terapia medicamentosa (véase más adelante Síntomas neuropsiquiátricos, incluyendo manía y trastorno bipolar; Efectos colaterales). Se debe considerar la realización de un cambio en el régimen terapéutico, incluyendo la posible suspensión de la terapia medicamentosa, en aquellos pacientes que experimenten agravamiento clínico (incluyendo el desarrollo de nuevos síntomas) y/o el surgimiento de ideación/comportamiento suicida, especialmente si estos síntomas son graves, de aparición abrupta, o si no formaban parte de los síntomas que ya presentaba el paciente. Síntomas neuropsiquiátricos, incluyendo manía y trastorno bipolar: Se han reportado síntomas neuropsiquiátricos (véase Efectos colaterales). En particular, se ha observado sintomatología psicótica y maníaca, principalmente en pacientes con antecedentes de enfermedades psiquiátricas. Además, existe la posibilidad de que los pacientes experimenten un episodio depresivo mayor como la presentación de trastorno bipolar. Es de consenso general (aunque no se estableció en los estudios controlados) que al tratar un episodio de este tipo con algún agente antidepresivo solo, es posible que aumente la probabilidad de una precipitación de algún episodio mixto/maníaco en los pacientes en riesgo de padecer trastorno bipolar. La limitada información clínica existente sobre el uso de la anfebutamona (bupropión) en combinación con estabilizadores del humor, en pacientes con antecedentes de trastorno bipolar, sugiere una tasa reducida de cambio a la fase maníaca. Antes de iniciar el tratamiento con algún antidepresivo, se debe evaluar adecuadamente a los pacientes, con el fin de determinar si se encuentran en riesgo de presentar trastorno bipolar; estas pruebas de detección deben incluir un historial psiquiátrico detallado, incluyendo un historial familiar de suicidio, trastorno bipolar y depresión. Enfermedad cardiovascular: Existe poca experiencia clínica sobre el uso de la anfebutamona (bupropión) para tratar la depresión en pacientes con enfermedades cardiovasculares. Se debe tener cuidado al emplear Wellbutrin XL en estos pacientes. Sin embargo, la anfebutamona (bupropión) fue generalmente bien tolerado, en estudios para cesación de tabaquismo realizados en pacientes con cardiopatía isquémica (véase Estudios Clínicos). Presión arterial: En un estudio realizado en sujetos no deprimidos (incluyendo tanto fumadores como no fumadores), que padecían hipertensión en Etapa I sin tratamiento, la anfebutamona (bupropión) no produjo efecto estadísticamente significativo en la presión arterial. Sin embargo, se han recibido comunicaciones espontáneas de aumentos (en ocasiones graves) en la presión arterial (véase Efectos colaterales); además, el uso concomitante de Bupropión con algún sistema transdérmico de nicotina podría ocasionar elevaciones en la presión arterial (véase Interacciones). No existe información respecto a la seguridad de uso de anfebutamona clorhidrato en pacientes con infarto al miocardio reciente o enfermedad cardíaca inestable. Agitación e insomnio: En 3 estudios clínicos controlados con placebo de trastorno afectivo estacional con Wellbutrin XL, la incidencia de agitación, ansiedad e insomnio, se muestran en la tabla Nş1.
Ver Tabla
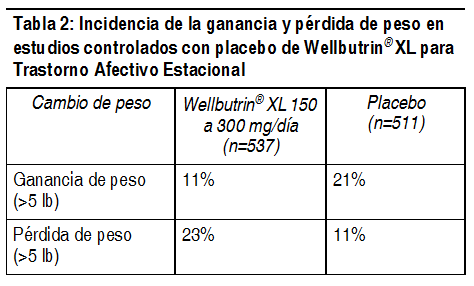
Alteración de peso y apetito: En 3 ensayos clínicos controlados con placebo de trastornos afectivo estacional con Wellbutrin® XL, el porcentaje de pacientes con ganancia o pérdida de peso, se muestran en la Tabla Nş2.
Ver Tabla
|
|
Precauciones:
Embarazo y lactancia: Embarazo: Fertilidad: No hay datos acerca del efecto de bupropión en la fertilidad humana. Un estudio de reproducción en ratas no reveló evidencia de disminución en la fertilidad (véase Datos Preclínicos de Seguridad). Embarazo: En algunos estudios epidemiológicos acerca del desenlace del embarazo, se ha reportado aumento en el riesgo de algunas malformaciones cardiovasculares en asociación con la exposición materna a bupropión durante el primer trimestre. Estos hallazgos no son consistentes en todos los estudios. El médico prescriptor necesitará sopesar la opción de tratamientos alternativos para mujeres embarazadas o que planeen embarazarse, y solo deberían prescribir Wellbutrin XL si los beneficios esperados son más importantes que los riesgos potenciales. La proporción de defectos cardiacos congénitos observados prospectivamente en embarazos con exposición prenatal a bupropión durante el primer trimestre en el Registro Internacional de Embarazo fue 9/675 (1.3%). En un estudio retrospectivo, realizado en una base de datos de cuidados de la salud administrados (n=7.005 lactantes), no se observó una mayor proporción de malformaciones congénitas (2.3%) o cardiovasculares (1.1%), asociadas con la exposición al bupropión durante el primer trimestre de embarazo (n=1.213 lactantes), en comparación con el uso de otros antidepresivos durante el primer trimestre (n=4.743 lactantes: 2.3% y 1.1% para malformaciones congénitas y cardiovasculares, respectivamente), o con el uso de bupropión fuera del primer trimestre (n=1.049: 2.2% y 1.0%, respectivamente). En un análisis retrospectivo de control de casos usando datos del Estudio Nacional de Prevención de Defectos Congénitos (National Birth Defects Prevention Study), hubo 12.383 lactantes identificados como casos y 5.869 lactantes identificados como controles. Se observó una asociación estadísticamente significativa entre la ocurrencia de un defecto del tracto de salida del ventrículo izquierdo en el lactante y el reporte hecho por las propias madres acerca del consumo de bupropión tempranamente durante el embarazo (n=10; OR ajustado =2.6; 95% IC 1.2 - 5.7). No se observó asociación entre el uso de bupropión por las madres y cualquier otro tipo de defecto cardíaco o con todas las categorías de defectos cardíacos combinados. Un análisis extendido de datos de control de casos del Estudio de Defectos Congénitos del Centro Epidemiológico Slone (Slone Epidemiology Center Birth Defects Study) incluyó 7.913 casos de lactantes con defectos cardíacos y 8.611 controles. No se encontró aumento estadísticamente significativo del defecto de salida del tracto izquierdo del corazón con el uso de bupropión de las madres (n=2; OR ajustado = 0.4; 95% IC 0.1 - 1.6). Sin embargo, se observó una asociación estadísticamente significativa de defectos septales ventriculares (n=17; OR ajustado =2.5; 95% IC 1.3 - 5.0) después del uso de bupropión solo durante el primer trimestre. Lactancia: Como la anfebutamona (bupropión) y sus metabolitos se excretan en la leche materna, se debe aconsejar a las madres que no amamanten a sus hijos mientras estén bajo tratamiento con Wellbutrin XL. Efectos sobre la capacidad para conducir y operar maquinaria: Al igual que otros fármacos que actúan en el sistema nervioso central (SNC), es posible que el bupropión afecte la capacidad de desempeńar tareas que requieren juicio o habilidades psicomotoras y cognitivas. Por tanto, los pacientes deberán tener cuidado antes de conducir o utilizar maquinaria, hasta que estén razonablemente seguros de que Wellbutrin XL comprimidos no los afecta de manera adversa en su desempeńo.
|
|
Interacciones Medicamentosas:
La anfebutamona (bupropión) se metaboliza a su metabolito activo principal, el hidroxibupropión, mediante la isoenzima IIB6 del citocromo P450 (CYP2B6), principalmente (véase Farmacocinética). Por tanto, se debe tener cuidado al coadministrar Wellbutrin® XL con fármacos que se sabe afectan la isoenzima CYP2B6 (por ejemplo, orfenadrina, ciclofosfamida, ifosfamida, ticlopidina, clopidogrel). Aunque el bupropión no se metaboliza a través de la isoenzima CYP2D6, los estudios in vitro realizados en el P450 humano demostraron que el bupropión y el hidroxibupropión son inhibidores de la vía CYP2D6. En un estudio farmacocinético realizado en humanos, la administración de bupropión aumentó las concentraciones plasmáticas de desipramina. Este efecto tuvo una duración de cuando menos 7 días después de administrar la última dosis de bupropión. La terapia concomitante con fármacos metabolizados predominantemente por esta isoenzima (como ciertos beta-bloqueantes, antiarrítmicos, IRSSs, ATCs, antipsicóticos), debe iniciarse a la dosis más baja del rango de dosificación de la medicación concomitante. Si se adiciona Wellbutrin® XL al régimen de tratamiento de un paciente que ya está recibiendo algún medicamento metabolizado por la isoenzima CYP2D6, deberá considerarse la necesidad de reducir la dosificación del medicamento original, especialmente de aquellos medicamentos concomitantes con un índice terapéutico estrecho (véase Farmacocinética). Fámacos que requieren activación metabólica a través de CYP2D6 para ser efectivos (por ejemplo tamoxifeno), podrían tener reducida su eficacia cuando son administrados concomitantemente con inhibidores de CYP2D6 tal como bupropión. Estudios realizados en animales demostraron un aumento en la toxicidad aguda asociada a la anfebutamona cuando se administra fenelzina. Aunque el citalopram (un ISRS) no se metaboliza principalmente a través de la isoenzima CYP2D6, en un estudio el bupropión aumentó la Cmáx y el ABC del citalopram en un 30% y 40%, respectivamente. Como el bupropión se metaboliza ampliamente, la administración concomitante de fármacos inductores metabólicos (por ejemplo, carbamazepina, fenobarbitona, fenitoína, ritonavir, efavirenz), o inhibidores metabólicos, podría afectar su actividad clínica. En una serie de estudios realizados en voluntarios sanos, la administración de ritonavir (100 mg 2 veces al día ó 600 mg 2 veces al día), o 100 mg de ritonavir más 400 mg de lopinavir (Kaletra®) 2 veces al día, redujo el nivel de exposición al bupropión y sus metabolitos principales de una manera proporcional a la dosis, en aproximadamente 20 a 80%. Similarmente, efavirenz 600 mg 1 vez al día durante 2 semanas redujo la disponibilidad de bupropión en aproximadamente 55%. Se cree que este efecto de ritonavir/Kaletra® y efavirenz se debe a la inducción del metabolismo del bupropión. Los pacientes que reciben tratamiento con cualquiera de estos fármacos con bupropión podrían requerir mayores dosis de bupropión, pero no se deberá exceder la dosis máxima recomendada de bupropión. Aunque los datos clínicos no identifican alguna interacción farmacocinética entre el bupropión y el alcohol, se han hecho reportes poco frecuentes de eventos neuropsiquiátricos adversos, o reducción en la tolerancia al alcohol, en pacientes que beben alcohol durante el tratamiento con bupropión. Se debe minimizar o evitar el consumo de alcohol durante el tratamiento con Wellbutrin® XL. La limitada información clínica sugiere una mayor incidencia de eventos adversos neuropsiquiátricos en pacientes que reciben tratamiento con bupropión en forma concomitante con levodopa o amantadina. Se debe tener precaución al administrar Wellbutrin® XL a pacientes que reciban tratamiento concurrente con levodopa o amantadina. La administración de dosis orales múltiples de bupropión no produjo efectos estadísticamente significativos en la farmacocinética de la lamotrigina, administrada en dosis únicas en 12 sujetos. Además, sólo aumentó ligeramente el ABC del glucurónido de lamotrigina. Existe la posibilidad de que el uso concomitante de Wellbutrin® XL y algún sistema transdérmico de nicotina (NTS, por sus siglas en inglés), ocasionen elevaciones en la presión arterial. Interacciones involucrando pruebas de laboratorio: Se ha reportado que Wellbutrin XL interfiere con algunas pruebas rápidas en orina que pueden resultar en lecturas falsas positivas, particularmente para anfetaminas. Se debe considerar un método químico alternativo más específico para confirmar el resultado positivo.
|
|
Sobredosificación:
Síntomas y signos: Además de aquellos efectos comunicados como efectos colaterales, los casos de sobredosificación han provocado síntomas que incluyen somnolencia, pérdida de la conciencia y cambios en el ECG, como trastornos de la conducción (incluyendo prolongación del intervalo QRS) o arritmias. Se han comunicado casos de ingestión aguda de dosis que exceden 10 veces a la terapéutica máxima. Tratamiento: En caso de una sobredosis, se recomienda la hospitalización. Se debe vigilar el ECG y los signos vitales. Asegúrese de que la permeabilidad de la vía aérea, la oxigenación y la ventilación sean adecuadas. Se recomienda el uso de carbón activado. No se conoce algún antídoto específico para la anfebutamona (bupropión). El tratamiento ulterior deberá ser el indicado por el médico o el recomendado por el centro nacional de toxicología, donde esté disponible.
|
|
Incompatibilidades:
Ninguna comunicada.
|
|
Conservación:
Vida útil: La fecha de caducidad se indica en el empaque. Precauciones especiales de almacenamiento: No almacenar a temperaturas superiores a 25ş C.
|
|



{kind=link}
{kind=link}
{kind=link}