 |
Descripción:
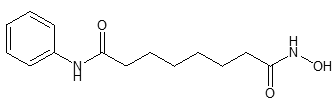
Cápsulas de 100 mg, de gelatina dura, blancas y opacas, con "568" impreso sobre "100 mg" con tinta negra en el código de barras radial del cuerpo de la cápsula. Zolinza contiene vorinostat, cuya descripción química es N-hidroxi-N'-feniloctanodiamido. Su fórmula empírica es C14H20N2O3. Su peso molecular es 264.32; y la fórmula estructural es:
Ver Tabla
El vorinostat es un polvo blanco a naranja claro. Es muy levemente soluble en agua, levemente soluble en etanol, isopropanol y acetona, fácilmente soluble en dimetilsulfóxido e insoluble en cloruro de metileno. No tiene centros quirales y no es higroscópico. La calorimetría diferencial de barrido osciló entre 161.7 (endotérmico) y 163.9°C. El pH de las soluciones del principio activo vorinostat saturado en agua fue 6.6. Se determinó que el pKa del vorinostat es 9.2. Cada cápsula de Zolinza 100 mg para administración oral contiene 100 mg de vorinostat y los siguientes excipientes: celulosa microcristalina, croscarmelosa sódica y estearato de magnesio. Los excipientes de la cápsula y su recubrimiento son dióxido de titanio, gelatina, tinta negra.
|
|
Indicaciones:
Zolinza está indicado para el tratamiento de manifestaciones cutáneas en pacientes con linfoma cutáneo de células T (CTCL por sus siglas en inglés) cuya enfermedad sea progresiva, persistente o recurrente durante o después del tratamiento con 2 terapias sistémicas.
|
|
Propiedades:
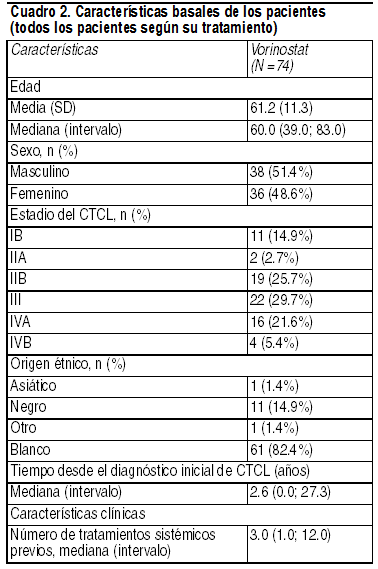
Farmacología clínica: Mecanismo de acción: Vorinostat inhibe la actividad enzimática de las histonas deacetilasas HDAC1, HDAC2 y HDAC3 (Clase I); y HDAC6 (Clase II) en concentraciones nanomolares (IC50< 86 nM). Estas enzimas catalizan la eliminación de grupos acetilo de los residuos de lisina de proteínas, incluidas las histonas y los factores de transcripción. En algunas células cancerosas, hay una sobreexpresión de las HDAC o una recuperación aberrante de las HDAC a factores de transcripción oncogénicos que causan la hipoacetilación de las histonas nucleosomales centrales. La hipoacetilación de las histonas está asociada a una estructura de cromatinas condensada y a la represión de la transcripción genética. Inhibir la actividad de las HDAC permite la acumulación de grupos acetilos en los residuos de lisina de las histonas, lo que causa una estructura de cromatina abierta y la activación de la transcripción. In vitro, el vorinostat provoca la acumulación de histonas acetiladas e induce la detención del ciclo celular y/o la apoptosis de algunas células transformadas. El mecanismo del efecto antineoplásico del vorinostat no se ha descrito por completo. Farmacocinética. Absorción: Se evaluó la farmacocinética de vorinostat en 23 pacientes con recidiva del cáncer o cáncer refractario en etapa avanzada. Tras la administración por vía oral de una dosis única de 400 mg de vorinostat junto con una comida rica en grasas, el promedio ± la desviación estándar del área bajo la curva (ABC), la concentración sérica máxima (Cmax) y la mediana (intervalo) del tiempo para alcanzar la concentración máxima (Tmax) fueron 5.5±1.8 µM · hr; 1.2±0.62 µM y 4 (2-10) horas, respectivamente. En ayunas, la administración por vía oral de una dosis única de 400 mg de vorinostat arrojó un promedio de ABC, Cmax y una mediana Tmax de 4.2±1.9 µM · hr; 1.2±0.35 µM y 1.5 (0,5-10) horas, respectivamente. Por lo tanto, administrar el vorinostat por vía oral junto con una comida rica en grasas causó un aumento (33%) del nivel de absorción y una disminución moderada de la velocidad de absorción (la Tmax se retrasó 2.5 horas), al compararlo con su administración en ayunas. Si embargo, no se anticipa que estos pequeńos efectos sean clínicamente importantes. En los estudios clínicos conducidos en pacientes con CTCL, el vorinostat se ingirió con alimentos. En estado estacionario, la administración oral de múltiples dosis de 400 mg de vorinostat junto con alimentos arrojó un promedio de ABC, Cmax y una Tmax mediana de 6.0±2.0 µM · hr; 1.2±0.53 µM y 4 (0,5-14) horas, respectivamente. Distribución: El vorinostat se une en aproximadamente un 71% a las proteínas plasmáticas humanas a concentraciones de entre 0.5 y 50 µg/ml. Metabolismo: Las vías principales del metabolismo de vorinostat involucran la glucuronidación e hidrólisis, seguidas por la ß-oxidación. Se midieron los niveles séricos de 2 metabolitos en seres humanos: O-glucurónido de vorinostat, y ácido 4-anilino- 4-oxobutanoico. Ambos metabolitos son farmacológicamente inactivos. Comparadas con vorinostat, la media de las exposiciones séricas de estado estacionario en seres humanos al O-glucurónido de vorinostat y al ácido 4-anilino-4-oxobutanoico fueron 4 y 13 veces más altas, respectivamente. Estudios in vitro conducidos en microsomas hepáticos humanos indican que la biotransformación por citocromas P450 (CYP) es muy mínima. Excreción: El vorinostat se elimina principalmente por metabolismo, con una recuperación inferior a 1% de la dosis en forma del fármaco íntegro en la orina, lo cual indica que la excreción renal no participa en su eliminación. La recuperación media en la orina de 2 metabolitos farmacológicamente inactivos en estado estacionario fue 16±5.8% de la dosis de vorinostat en forma de O-glucurónido de vorinostat, y 36±8.6% de la dosis del fármaco en forma de ácido 4-anilino-4-oxobutanoico. La recuperación total del vorinostat y los 2 metabolitos en la orina promedió un 52±13.3% de la dosis de vorinostat. La semivida terminal (t˝) media fue aproximadamente 2.0 horas para el vorinostat y el metabolito O-glucurónido, mientras que la del metabolito ácido 4-anilino-4-oxobutanoico fue de 11 horas. Poblaciones especiales: Basado en un análisis exploratorio de datos limitados, el sexo, la raza y la edad no parecen tener efectos significativos sobre la farmacocinética del vorinostat. Pediátrica: El vorinostat no ha sido evaluado en pacientes menores de 18 ańos. Insuficiencia hepática: El vorinostat no ha sido evaluado en pacientes con insuficiencia hepática (consulte Uso en Poblaciones Específicas). Insuficiencia renal: El vorinostat no ha sido evaluado en pacientes con insuficiencia renal. Sin embargo, la excreción renal no participa en la eliminación del vorinostat (consulte Uso en Poblaciones Específicas). Efectos farmacocinéticos de vorinostat administrado con otros fármacos: En la Cmax de estado estacionario correspondiente a la dosis de 400 mg (Cmax de 1.2 µM frente a IC50 de >75 µM), el vorinostat no inhibe las enzimas CYP metabolizadoras de fármacos de los microsomas hepáticos humanos. Estudios de expresión génica conducidos en hepatocitos humanos detectaron que el vorinostat tiene cierto potencial para suprimir la actividad de las CYP2C9 y CYP3A4, al administrarlo en concentraciones superiores ( ł 10 µM) a las de interés farmacológico. Por lo tanto, no se anticipa que el vorinostat afecte la farmacocinética de otros fármacos. Dado que el vorinostat no se elimina por las vías de CYP, se espera que no esté sujeto a interacciones entre fármacos al administrarlo concomitantemente con fármacos de actividad inhibitoria o inductora de CYP comprobada. Sin embargo, no se han realizado estudios clínicos formales para evaluar interacciones farmacológicas con vorinostat. Toxicología no clínica: Carcinogénesis, mutagénesis, alteración de la fertilidad: No se han realizado estudios de carcinogénesis con vorinostat. El vorinostat fue mutagénico in vitro en los ensayos de mutación reversa bacteriana (test de Ames), causó aberraciones cromosómicas in vitro en células de ovario de hámster chino (CHO), e incrementó la incidencia de micronúcleos en eritrocitos al administrarlo a ratones (ensayo de micronúcleos en ratones). Se identificaron efectos sobre el aparato reproductor femenino en el estudio sobre fertilidad tras la administración por vía oral, en el que las hembras recibieron el fármaco a partir de 14 días antes del apareamiento y hasta el 7ş día de gestación. En ratas, las dosis de 15, 50 y 150 mg/kg/día causaron exposiciones de aproximadamente 0.15; 0.36 y 0.70 veces la exposición clínica anticipada, basada en el ABC. Se observaron aumentos dosis-dependientes de los cuerpos lúteos con dosis ł 15 mg/kg/día, lo que resultó en el incremento de las pérdidas en la peri-implantación con dosis ł 50 mg/kg/día. Con la dosis de 150 mg/kg/día, se observó un incremento de las muertes fetales y reabsorciones. No se observaron efectos sobre el desempeńo reproductivo en las ratas macho que recibieron el fármaco (dosis de 20, 50 y 150 mg/kg/día; exposiciones aproximadas de 0.15; 0.36 y 0.70 veces la exposición clínica esperada basada en el ABC) durante los 70 días previos a su apareamiento con hembras no tratadas (consulte Advertencias y Precauciones). Estudios clínicos: Linfoma cutáneo de células T: En 2 estudios clínicos abiertos, se ha evaluado a pacientes con CTCL refractario para determinar su tasa de respuesta a Zolinza de administración oral. Uno de los estudios clínicos se realizó con un único grupo de tratamiento, y en el otro se evaluaron varios regímenes de administración. En ambos estudios, se trató a los pacientes hasta la progresión de la enfermedad o hasta que presentaron toxicidad intolerable. Estudio 1: En un estudio multicéntrico, no randomizado, abierto y de un solo grupo de tratamiento, 74 pacientes con CTCL en etapa avanzada fueron tratados con una dosis de 400 mg de Zolinza, administrada 1 vez al día. El criterio primario de valoración fue la tasa de respuesta a Zolinza de administración oral, en el tratamiento del cáncer de piel en pacientes con CTCL en etapa avanzada (estadio IIB y superior) cuya enfermedad presentó avanzó, persistió o presentó una recidiva durante o después del tratamiento con 2 terapias sistémicas. Los pacientes enrolados debían haber recibido, o bien presentar intolerancia o no ser aptos para recibir, bexaroteno. Los investigadores evaluaron cuantitativamente la extensión del cáncer, con una herramienta de evaluación de la severidad ponderada (SWAT, por su sigla en inglés). El investigador midió el porcentaje de la superficie corporal total (%TBSA) afectada, separado por lesiones de parche, placas y tumores en 12 zonas del cuerpo, usando la palma del paciente como "regla". El %TBSA total de cada tipo de lesión se multiplicó por un factor de ponderación de severidad (1=parche, 2=placa y 4=tumor) y calculó para derivar el puntaje SWAT. La eficacia se midió como respuesta clínica completa (CCR, por su sigla en inglés), definida como ausencia de evidencia de la enfermedad; o respuesta parcial (PR, por su sigla en inglés), definida como una disminución ł 50% en el puntaje SWAT de evaluación de la piel respecto del puntaje basal. Tanto la CCR como la PR debían mantenerse durante al menos 4 semanas. Los criterios secundarios de la eficacia incluyeron la duración de la respuesta, el tiempo transcurrido hasta el avance de la enfermedad y el tiempo hasta la observación de una respuesta objetiva. La población había sido expuesta a una mediana de tres terapias previas (intervalo de 1 a 12). El Cuadro 2 resume las características demográficas y de la enfermedad correspondientes a la población del Estudio 1.
Ver Tabla
La tasa global de respuesta objetiva fue 29.7% (22/74, IC de 95% [19.7 a 41.5%]) en todos los pacientes tratados con Zolinza. En los pacientes con CTCL estadio IIB y superior, la tasa global de respuesta objetiva fue 29.5% (18/61). Un paciente con CTCL estadio IIB alcanzó una CCR. Las medianas del tiempo a la respuesta fueron 55 y 56 días (intervalo de 28 a 171 días), respectivamente, en la población completa y en los pacientes con CTCL estadio IIB y superior. No obstante, en algunos casos infrecuentes los pacientes tardaron hasta 6 meses en presentar una respuesta objetiva a Zolinza. No se obtuvo la mediana de duración de la respuesta, dado que la mayoría de las respuestas se mantenía al momento del análisis; sin embargo, se calcula que superó los 6 meses tanto en la población general, como en la de pacientes con CTCL estadio IIB y superior. Al definir el término de la respuesta como un aumento de 50% en el puntaje SWAT respecto del puntaje valle, la mediana estimada de duración de la respuesta fue 168 días, y la mediana de tiempo transcurrido hasta la progresión del tumor fue 202 días. Al usar un aumento de 25% en el puntaje SWAT respecto del puntaje valle como criterio para determinar la progresión del tumor, la mediana estimada del tiempo a la progresión fue 148 días para la población completa y 169 días en los 61 pacientes con CTCL estadio IIB y superior. La respuesta a cualquiera de las terapias sistémicas previas no parece ser predictiva de la respuesta a Zolinza. Estudio 2: En un estudio no randomizado y abierto, se evaluó Zolinza para determinar la tasa de respuesta en pacientes con CTCL refractario o intolerante a por lo menos un tratamiento. En este estudio, 33 pacientes fueron asignados a uno de 3 cohortes: la cohorte 1, con 400 mg 1 vez al día; la cohorte 2, con 300 mg 2 veces al día, 3 días a la semana; o la cohorte 3, con 300 mg 2 veces al día durante 14 días, seguidos de un reposo de 7 días (inducción). En la cohorte 3, si no se observaba por lo menos una respuesta parcial, se administraba a los pacientes un régimen de mantención de 200 mg 2 veces al día. El criterio primario de valoración de la eficacia, la respuesta objetiva, se midió con el instrumento de 7 ítems Escala global de evaluación del médico (PGA, por su sigla en inglés). El investigador evaluó la mejoría o el agravamiento de la enfermedad general en comparación con el estado basal, basándose en la impresión clínica global. También se evaluaron e incluyeron en la impresión clínica global las lesiones cutáneas de referencia y las no usadas como referencia, así como los tumores cutáneos, los ganglios linfáticos y todas las demás manifestaciones de la enfermedad. La CCR requería que el 100% de las observaciones vinculadas al cáncer hubieran desaparecido, y la PR requería una mejoría de por lo menos 50% en los hallazgos de la enfermedad. La mediana de edad fue 67.0 ańos (intervalo de 26.0 a 82.0). El 55% de los pacientes eran hombres; y el 45%, mujeres. El 15% de los pacientes tenía CTCL estadio IA, IB, o IIA; y el 85% restante padecía CTCL estadio IIB, III, IVA, o IVB. La mediana del número de terapias sistémicas previas era 4 (intervalo de 0,0 a 11,0). En el grupo de todos los pacientes tratados, se registró una respuesta objetiva en 24.2% (8/33) de la población general, en 25% (7/28) de los pacientes con cáncer de estadio IIB o superior; y en 36.4% (4/11) de los pacientes con síndrome de Sézary. Las tasas globales de respuesta fueron 30.8%; 9.1% y 33.3% en la cohorte 1, la cohorte 2 y la cohorte 3, respectivamente. El régimen de 300 mg 2 veces al día causó más toxicidad que el de 400 mg administrado 1 vez al día, y no exhibió beneficios clínicos adicionales. No se observó CCR. Entre los 8 pacientes que respondieron al tratamiento del estudio, la mediana del tiempo transcurrido hasta la respuesta fue 83.5 días (intervalo de 25 a 153 días). La mediana de duración de la respuesta fue 106 días (intervalo de 66 a 136 días). La mediana del tiempo hasta la progresión fue 211.5 días (intervalo de 94 a 255 días).
|
|
Posología:
Información de dosificación: La dosis recomendada es 400 mg por vía oral 1 vez al día, con alimentos. El tratamiento puede mantenerse mientras no exista evidencia de progreso de la enfermedad o de toxicidad inaceptable. Las cápsulas de Zolinza no deben abrirse ni aplastarse (consulte Forma de suministro/Almacenamiento y Manipulación). Modificaciones a la dosis: Si el paciente no tolera la terapia, la dosis puede reducirse a 300 mg por vía oral 1 vez al día, con alimentos. De ser necesario, la dosis podría reducirse aún más, a 300 mg 1 vez al día con alimentos, durante 5 días consecutivos cada semana. Dosificación en poblaciones especiales: No hay información disponible sobre el fármaco en pacientes con insuficiencia renal o hepática (consulte Farmacocinética).
|
|
Modo de Empleo:
Se deberá considerar los procedimientos de manipulación y eliminación adecuados para los antineoplásicos. Se han publicado diversas directrices sobre este tema. No existe consenso general respecto de que todos los procedimientos recomendados en tales directrices sean necesarios o adecuados. No se deben abrir ni aplastar las cápsulas de Zolinza (vorinostat). Se debe evitar que el polvo contenido en las cápsulas de Zolinza entre en contacto directo con la piel o las membranas mucosas. En caso de ocurrir tal contacto, lave bien el área afectada como se indica en las referencias. El personal deberá evitar la exposición a cápsulas aplastadas y/o rotas (consulte Toxicología no clínica).
|
|
Efectos Colaterales:
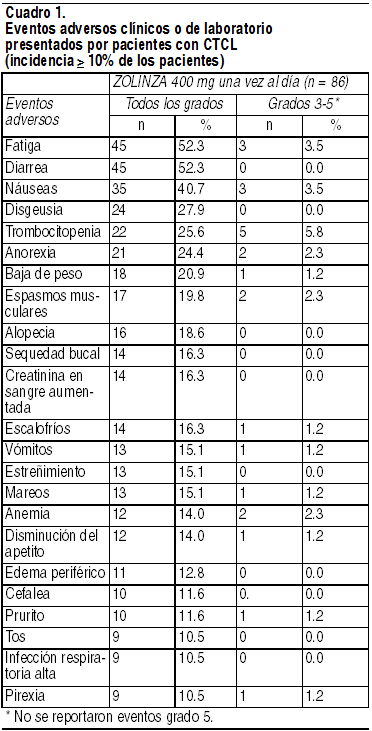
Las reacciones adversas más frecuentes relacionadas con el fármaco pueden clasificarse en 4 grupos de síntomas: síntomas gastrointestinales (diarrea, náusea, anorexia, pérdida de peso, vómitos, estreńimiento), síntomas constitucionales (fatiga, escalofríos), anomalías hematológicas (trombocitopenia, anemia), y trastornos del gusto (disgeusia, sequedad bucal). Las reacciones adversas serias más frecuentes relacionadas con el fármaco fueron embolia pulmonar y anemia. Experiencia en estudios clínicos: La seguridad de Zolinza se evaluó en 107 pacientes con CTCL, en 2 estudios clínicos de un único grupo de tratamiento en los que 86 pacientes recibieron 400 mg del fármaco 1 vez al día. Los datos presentados más abajo reflejan la exposición de los 86 pacientes a 400 mg de Zolinza, administrados 1 vez al día durante una media de 97.5 días de tratamiento (intervalo de 2 a 480+ días). 17 (19.8%) pacientes fueron expuestos durante más de 24 semanas, y 8 (9.3%) pacientes fueron expuestos durante más de 1 ańo. Los pacientes con CTCL que conformaron la población estudiada tenían entre 37 y 83 ańos de edad; se dividían en 47.7% de mujeres y 52.3% de hombres; y el 81.4% era blanco, el 16.3% era negro, y el 1.2% era asiático o de distintas razas. Debido a que los estudios clínicos se conducen en condiciones que varían ampliamente, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas registradas en estudios clínicos de otro fármaco, y podrían no reflejar las tasas observadas en la práctica. Reacciones adversas comunes: En el Cuadro 1 se resume la frecuencia con la que pacientes con CTCL experimentaron eventos adversos específicos, independientemente de la causalidad, de acuerdo con los Criterios de Terminología Común para los Eventos Adversos del Instituto Nacional (estadounidense) del Cáncer (NCI CTCAE, versión 3.0).
Ver Tabla
La frecuencia de las reacciones más severas de trombocitopenia, anemia (consulte Advertencias y precauciones) y fatiga aumentó al administrar dosis de Zolinza superiores a 400 mg 1 vez al día. Reacciones adversas serias: Los eventos adversos serios observados con más frecuencia en los 86 pacientes con CTCL de 2 estudios clínicos, independientemente de la relación de causalidad con el fármaco, fueron embolia pulmonar, reportada en 4.7% (4/86) de los pacientes; carcinoma escamoso, reportado en 3.5% (3/86) de los pacientes; y anemia, reportada en 2.3% (2/86) de ellos. Hubo eventos únicos de colecistitis, muerte (por causa desconocida), trombosis venosa profunda, infección enterocócica, dermatitis exfoliativa, hemorragia gastrointestinal, infección, neumonía lobar, infarto de miocardio, accidente cerebrovascular isquémico, obstrucción de la unión urétero-pélvica, sepsis, lesión a la médula espinal, bacteriemia estreptocócica, síncope, linfoma de células T, trombocitopenia y obstrucción ureteral. Suspensión del tratamiento: De los pacientes con CTCL que recibieron la dosis diaria de 400 mg, el 9.3% (8/86) descontinuó el tratamiento con Zolinza debido a eventos adversos. Tales eventos adversos, independientemente de su relación de causalidad con el fármaco, incluyeron anemia, edema angioneurótico, astenia, dolor torácico, dermatitis exfoliativa, muerte, trombosis venosa profunda, accidente cerebrovascular isquémico, letargo, embolia pulmonar y lesión a la médula espinal. Modificaciones a la dosis: De los pacientes con CTCL que recibieron 400 mg de Zolinza administrados 1 vez al día, fue necesario modificarle la dosis al 10.5% (9/86) debido a eventos adversos. Tales eventos adversos incluyeron aumento de la creatinina sérica, disminución del apetito, hipopotasemia, leucopenia, náuseas, neutropenia, trombocitopenia y vómitos.El tiempo promedio transcurrido hasta la presentación del primer evento adverso que resultó en la reducción de la dosis fue 42 días (intervalo de 17 a 263 días). Anomalías de laboratorio: Se reportaron anomalías de laboratorio en (todos) los 86 pacientes con CTCL que recibieron la dosis de 400 mg 1 vez al día. Se reportó el aumento de la glucosa sérica como anomalía de laboratorio en el 69% (59/86) de los pacientes con CTCL que recibieron la dosis de 400 mg en una administración diaria; pero sólo 4 de estas anomalías fueron graves (grado 3). Se reportó el aumento de la glucosa sérica como evento adverso en el 8.1% (7/86) de los pacientes con CTCL que recibieron la dosis de 400 mg 1 vez al día [consulte Advertencias y Precauciones]. Se detectaron aumentos temporales de creatinina sérica en el 46.5% (40/86) de los pacientes con CTCL que recibieron la dosis de 400 mg 1 vez al día. Según los criterios NCI CTCAE, 34 de tales anomalías de laboratorio fueron grado 1, 5 fueron grado 2, y 1 fue grado 3. Se detectó proteinuria como anomalía de laboratorio en 38 de 74 (51.4%) pacientes sometidos a análisis. Se desconoce cuál es la importancia clínica de este descubrimiento. Deshidratación: Dados los reportes en los que se ha mencionado la deshidratación como evento adverso serio relacionado con el medicamento en estudios clínicos, se le dió a los pacientes la instrucción de beber por lo menos 2 litros diarios de líquido, para hidratarse adecuadamente (consulte Advertencias y Precauciones). Reacciones adversas en pacientes que no sufren CTCL: Las frecuencias de eventos adversos individuales fueron considerablemente más altas en la población que no padecía CTCL. Los eventos adversos serios relacionados con el medicamento que se reportaron en la población que no padecía CTCL y no en la población con CTCL incluyeron un único evento de visión borrosa, astenia, hiponatremia, hemorragia tumoral, síndrome de Guillain-Barré, insuficiencia renal, retención urinaria, tos, hemoptisis, hipertensión y vasculitis.
|
|
Contraindicaciones:
Ninguna.
|
|
Advertencias:
Tromboembolismo: Dado que se han reportado casos de embolia pulmonar y trombosis venosa profunda como reacciones adversas, los médicos deberán estar alerta a los signos y síntomas de estos eventos, especialmente en el caso de pacientes con antecedentes de eventos tromboembólicos (consulte Efectos Colaterales). Hematológicas: El tratamiento con Zolinza puede causar trombocitopenia y anemia dosis-dependientes. Si los recuentos plaquetarios y/o la hemoglobina disminuyen durante el tratamiento con Zolinza, se deberá modificar la dosis o suspender el tratamiento (consulte Posología, Advertencias y Precauciones y Efectos Colaterales). Gastrointestinales: Se han reportado alteraciones gastrointestinales, incluidas náuseas, vómitos y diarrea (consulte Efectos Colaterales); las que podrían requerir la administración de antieméticos y antidiarreicos. Se deberá restituir los líquidos y electrolitos para prevenir la deshidratación (consulte Efectos colaterales). Las náuseas, vómitos y diarrea preexistentes deberán controlarse adecuadamente antes de comenzar la terapia con Zolinza. Hiperglucemia: Se ha observado hiperglucemia en pacientes que han recibido Zolinza (consulte Efectos colaterales). Se deberá monitorear la glucosa sérica, especialmente en pacientes diabéticos o potencialmente diabéticos. Podría ser necesario ajustar la alimentación y/o el tratamiento en caso de observarse un aumento de la glucemia. Prolongación del intervalo QTc: No se ha llevado a cabo un estudio definitivo del efecto de vorinostat sobre el intervalo QTc. Tres de 86 pacientes con CTCL expuestos a una dosis de 400 mg 1 vez al día, registraron prolongación del intervalo QTc como evento adverso clínico grado 1 (> 450-470 mseg) o 2 (> 470-500 mseg o aumento > 60 mseg por sobre el valor basal). En un análisis retrospectivo de 3 estudios fase I y 2 estudios fase II, 116 pacientes fueron sometidos a un ECG basal y a por lo menos un ECG de seguimiento. Cuatro pacientes registraron una prolongación del intervalo QTc grado 2 (> 470-500 mseg, o aumento > 60 mseg por sobre el valor basal), y 1 paciente registró una prolongación del intervalo QTc grado 3 (> 500 mseg). De 49 pacientes sin CTCL incluidos en 3 estudios clínicos que recibieron una evaluación completa del intervalo QT, 2 registraron mediciones de QTc > 500 mseg y 1 registró una prolongación del QTc > 60 mseg. Monitoreo: Análisis de laboratorio: Los recuentos sanguíneos y análisis químicos, incluidos los electrolitos, la glucosa y la creatinina sérica, deberán monitorearse cuidadosamente cada 2 semanas durante los primeros 2 meses de tratamiento; y, posteriormente, 1 vez al mes. El monitoreo de electrolitos incluirá los niveles de potasio, magnesio y calcio. Se deberá realizar un ECG al inicio y periódicamente durante el tratamiento. Zolinza deberá administrarse con especial cuidado a los pacientes con síndrome de intervalo QT prolongado congénito, así como a quienes estén en tratamiento con antiarrítmicos u otros productos medicinales que causen prolongación del QT. Se deberá corregir la hipopotasemia o la hipomagnesemia antes de administrar Zolinza, así como considerar el monitoreo de los niveles de potasio y magnesio en pacientes sintomáticos (por ejemplo, pacientes con náuseas, vómitos, diarrea, desequilibrio hidroelectrolítico o síntomas cardíacos) [consulte Advertencias y Precauciones]. Otros Inhibidores de Histona Deacetilasa (HDAC): Se ha reportado trombocitopenia severa y hemorragia gastrointestinal con el uso concomitante de Zolinza y otros inhibidores de HDAC (por ejemplo, ácido valproico). Monitoree el recuento de plaquetas cada 2 semanas durante los primeros 2 meses (consulte Interacciones con otros Medicamentos). Embarazo: Categoría D de uso en embarazo. Zolinza puede causar dańo fetal al administrarlo a mujeres embarazadas. No se han realizado estudios adecuados ni debidamente controlados de Zolinza en mujeres embarazadas. Los resultados obtenidos en estudios en animales indican que el vorinostat cruza la placenta, y es detectable en el plasma fetal en niveles de hasta 50% de las concentraciones detectadas en la madre. Se evaluaron dosis de hasta 50 y 150 mg/kg/día en ratas y conejos, respectivamente (aproximadamente 0.5 veces la exposición aplicada a seres humanos, basado en el ABC0-24 hrs). Los efectos del tratamiento sobre el desarrollo embrionario/fetal incluyeron una baja del peso medio de los fetos vivos; osificación incompleta del cráneo, las vértebras torácicas, las esternebras y variaciones del esqueleto (costillas cervicales, costillas supernumerarias, variaciones del número de vértebras y del arco del sacro) en ratas, con la dosis más alta de vorinostat evaluada. Se observó una disminución del peso promedio de los fetos vivos y un aumento en la incidencia de osificación incompleta de los metacarpianos en los conejos que recibieron 150 mg/kg/día. El nivel de efecto no observable (NOEL) para estos hallazgos fue 15 y 50 mg/kg/día (< 0.1 veces la exposición aplicada a seres humanos, basado en el ABC) en ratas y conejos, respectivamente. En conejos, todos los grupos tratados con el fármaco presentaron un aumento dosis-dependiente en la incidencia de malformaciones de la vesícula, en comparación con el grupo de control paralelo. Si se utiliza este fármaco durante el embarazo, o si la paciente queda embarazada mientras lo toma, se le deberá advertir sobre el potencial riesgo para el feto.
|
|
Precauciones:
Uso en poblaciones específicas: Embarazo: Categoría D de embarazo (consulte las Advertencias y Precauciones). Madres lactantes: Se desconoce si este fármaco se excreta por la leche materna. Considerando que muchos fármacos se excretan por la leche materna y debido al potencial de que los bebés lactantes presenten reacciones adversas graves a Zolinza, se deberá decidir entre suspender la lactancia o suspender el fármaco, tomando en cuenta la importancia que el tratamiento con Zolinza tenga para la madre. Uso pediátrico: No se han determinado la seguridad ni la efectividad de Zolinza en pacientes pediátricos. Uso geriátrico: Del número total de pacientes con CTCL que participaron en los estudios (N=107), el 46% tenía 65 ańos, o más; mientras que el 15% tenía 75 o más ańos. No se observaron diferencias globales de seguridad ni de efectividad entre estos sujetos y los pacientes más jóvenes, y en los reportes de otras experiencias clínicas no se han identificado diferencias de respuesta entre los pacientes ancianos y los más jóvenes. No obstante, no puede descartarse que algunas personas de edad avanzada puedan tener una sensibilidad mayor al fármaco. Uso en pacientes con insuficiencia hepática: El vorinostat no ha sido evaluado en pacientes con insuficiencia hepática. Dado que el vorinostat se elimina principalmente por metabolismo, los pacientes con insuficiencia hepática deberán ser tratados cuidadosamente (consulte Farmacología clínica). Uso en pacientes con insuficiencia renal: El vorinostat no ha sido evaluado en pacientes con insuficiencia renal. Sin embargo, la excreción renal no participa en la eliminación del vorinostat. Los pacientes con insuficiencia renal preexistente deberán ser tratados cuidadosamente (consulte Farmacología clínica).
|
|
Interacciones Medicamentosas:
Anticoagulantes derivados de la cumarina: Se observó la prolongación del tiempo de protrombina (PT) y del índice internacional normalizado (INR) en los pacientes que recibieron Zolinza y anticoagulantes derivados de la cumarina de manera concomitante. Los médicos deberán monitorear cuidadosamente el PT y el INR de los pacientes a quienes se les esté administrando paralelamente Zolinza y derivados de cumarina. Otros inhibidores de HDAC: Se ha reportado trombocitopenia severa y hemorragia gastrointestinal con el uso concomitante de Zolinza y otros inhibidores de HDAC (por ejemplo, ácido valproico). Monitoree el recuento de plaquetas cada 2 semanas durante los primeros 2 meses (consulte Advertencias y precauciones).
|
|
Sobredosificación:
No hay disponibilidad de información específica sobre el tratamiento de la sobredosis con Zolinza. En el caso de sobredosis, es razonable recurrir a las medidas de apoyo habituales; por ejemplo, quitar el material no absorbido del tracto digestivo, realizar monitoreo clínico, e implementar el tratamiento sintomático, de ser necesario. Se desconoce si el vorinostat es dializable.
|
|
Conservación:
Almacenar a no más de 30°C.
|
|
Observaciones:
Forma de suministro: Zolinza se presenta en cápsulas de gelatina dura, blancas y opacas, de 100 mg; con "568" sobre "100 mg" impreso dentro de una barra radial de tinta negra en el cuerpo de la cápsula.
|
|
Presentaciones:
Envase conteniendo 120 cápsulas.
|
|



{kind=link}
{kind=link}
{kind=link}